
The Virus–Host Interplay in Junín Mammarenavirus Infection



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Host–Virus Interactions Involved in JUNV Entry to the Cell
2.1. Transferrin Receptor 1 Is the Preferred Receptor for JUNV Entry
2.2. JUNV Alternatively Uses Noncanonical Receptors to Enter the Cell
2.3. JUNV Utilizes the Clathrin-Mediated Endocytic Pathway to Fulfill Viral Entry
2.4. JUNV Travels through Rab5-Early and Rab7-Late Endosomes before Membrane Fusion
2.5. TRIM-2 Protein Restricts JUNV Entry into the Cell
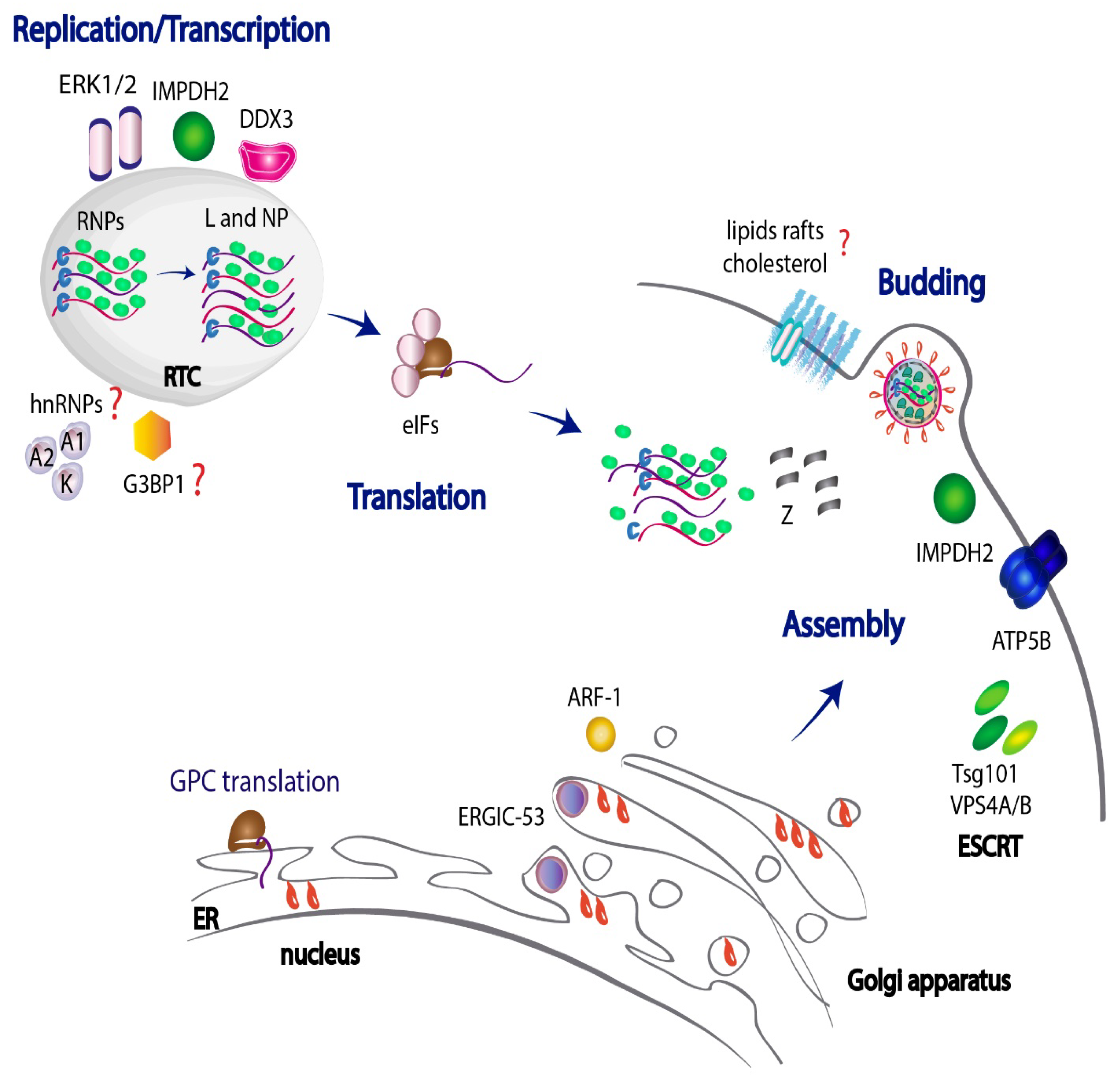
3. JUNV Genome Replication, Assembly and Budding
3.1. JUNV Replicates in Discrete Cytosolic Structures
3.2. Role of DEAD-Box RNA Helicase 3 (DDX3) in JUNV Replication
3.3. Host Kinases Impacting on JUNV RNA Synthesis
3.4. JUNV Z Protein-Binding Partners Related to the Cellular Energy Metabolism Support Virus Propagation
3.5. Host Heterogeneous Nuclear Ribonucleoproteins (HnRNPs) Are Sequestered by JUNV NP and Are Required for Viral Growth
3.6. Host Factors Involved in Regulation of JUNV Translation
3.7. JUNV Hijacks the Autophagy Pathway and Exploits Lipid-Droplets to Successfully Accomplish Viral Infection
3.8. Traffic of Viral Components within the Cell: Virus–Host Teamwork in the Assembly of the Glycoprotein Complex into Mature Virions
3.9. Cellular Targets Required for an Efficient Release of JUNV Particles
4. Host Immune Response upon JUNV Infection
4.1. RIG-I and MDA-5 Sense JUNV and Activate the IFN-I Cascade
4.2. Pathogenic and Nonpathogenic JUNV Strains Activate PKR Differently
4.3. JUNV Is Sensed by TLR Membrane Receptors in Mouse Macrophages
4.4. Interplay between JUNV and Several Host ISGs
4.5. JUNV NP and Z Proteins Modulate the IFN-Pathway
5. Reverse Genetics Systems as a Tool to Study JUNV Biology and Virus–Host Interactions
5.1. Rescue of Recombinant JUNV and Its Use in the Study of Virus Pathogenicity and Vaccine Development
5.2. Alternative Systems to Evaluate JUNV Replication, Assembly and/or Budding Processes
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Maiztegui, J.I. Clinical and epidemiological patterns of Argentine haemorrhagic fever. Bull World Health Organ. 1975, 52, 567–575. [Google Scholar] [PubMed]
- Maiztegui, J.I.; McKee, K.T., Jr.; Barrera Oro, J.G.; Harrison, L.H.; Gibbs, P.H.; Feuillade, M.R.; Enria, D.A.; Briggiler, A.M.; Levis, S.C.; Ambrosio, A.M.; et al. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J. Infect. Dis. 1998, 177, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, R.M.; Jaquenod de Giusti, C.; Sanchez Vallduvi, M.M.; Frik, J.; Ferrer, M.F.; Schattner, M. Junín virus. A XXI century update. Microbes Infect. 2011, 13, 303–311. [Google Scholar] [CrossRef]
- Maiztegui, J.I.; Fernandez, N.J.; de Damilano, A.J. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet 1979, 2, 1216–1217. [Google Scholar] [CrossRef]
- Enria, D.A.; Maiztegui, J.I. Antiviral treatment of Argentine hemorrhagic fever. Antivir. Res. 1994, 23, 23–31. [Google Scholar] [CrossRef]
- Romanowski, V.; Pidre, M.L.; Ferrelli, M.L.; Bender, C.; Gomez, R. Argentine hemorrhagic fever. In Viral Hemorrhagic Fevers; Singh, S.K., Ruzek, D., Eds.; Taylor & Francis Group: Abingdon, UK; CRC Press: Boca Raton, FL, USA, 2013; pp. 317–338, ISBN-13: 978-143-988-429-4, ISBN-10:143-988-429-3. [Google Scholar]
- Enria, D. Arenaviral hemorrhagic fevers: Argentine hemorrhagic fever and Lassa fever. In Emerging Neurological Infections; Power, C., Johnson, R.T., Eds.; Taylor & Francis Group: Abingdon, UK; CRC Press: Boca Raton, FL, USA, 2005; Volume 67, p. 505. ISBN 9780429142895. [Google Scholar]
- Charrel, R.N.; Coutard, B.; Baronti, C.; Canard, B.; Nougairede, A.; Frangeul, A.; Morin, B.; Jamal, S.; Schmidt, C.L.; Hilgenfeld, R.; et al. Arenaviruses and hantaviruses: From epidemiology and genomics to antivirals. Antivir. Res. 2011, 90, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Enria, D.A.; Briggiler, A.M.; Sánchez, Z. Treatment of Argentine hemorrhagic fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Kim, Y.J.; Venturini, V.; de la Torre, J.C. Progress in anti-Mammarenavirus drug development. Viruses 2021, 13, 1187. [Google Scholar] [CrossRef]
- Lavanya, M.; Cuevas, C.D.; Thomas, M.; Cherry, S.; Ross, S.R. siRNA screen for genes that affect Junín virus entry uncovers voltage-gated calcium channels as a therapeutic target. Sci. Transl. Med. 2013, 5, 204ra131. [Google Scholar] [CrossRef] [Green Version]
- Artuso, M.C.; Ellenberg, P.C.; Scolaro, L.A.; Damonte, E.B.; García, C.C. Inhibition of Junín virus replication by small interfering RNAs. Antivir. Res. 2009, 84, 31–37. [Google Scholar] [CrossRef]
- Rathbun, J.Y.; Droniou, M.E.; Damoiseaux, R.; Haworth, K.G.; Henley, J.E.; Exline, C.M.; Choe, H.; Cannon, P.M. Novel arenavirus entry inhibitors discovered by using a minigenome rescue system for high-throughput drug screening. J. Virol. 2015, 89, 8428–8443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, J.E.; Quintana, V.M.; Scolaro, L.A.; Castilla, V. Inhibitors of the p38 cell signaling pathway as antiviral compounds against Junín virus. Arch. Virol. 2022, 167, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, C.S.; García, C.C.; Damonte, E.B. Antiviral activity of A771726, the active metabolite of leflunomide, against Junín virus. J. Med. Virol. 2018, 90, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Riaño, E.; Ngo, N.; Devito, S.; Eggink, D.; Munger, J.; Shaw, M.L.; de la Torre, J.C.; Martínez-Sobrido, L. Inhibition of arenavirus by A3, a pyrimidine biosynthesis inhibitor. J. Virol. 2014, 88, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Han, Z.; Liu, Y.; Liu, W.; Lee, M.S.; Olson, M.A.; Ruthel, G.; Freedman, B.D.; Harty, R.N. A host-oriented inhibitor of Junin Argentine hemorrhagic fever virus egress. J. Virol. 2014, 88, 4736–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepúlveda, C.S.; García, C.C.; Levingston Macleod, J.M.; López, N.; Damonte, E.B. Targeting of arenavirus RNA synthesis by a carboxamide-derivatized aromatic disulfide with virucidal activity. PLoS ONE 2013, 8, e81251. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Cuevas, C.; Carocci, M.; Stubbs, S.H.; Ma, M.; Cureton, D.K.; Chao, L.; Evesson, F.; He, K.; Yang, P.L.; et al. Identification and characterization of a novel broad-Spectrum virus entry inhibitor. J. Virol. 2016, 90, 4494–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Gowen, B.B.; Juelich, T.L.; Sefing, E.J.; Brasel, T.; Smith, J.K.; Zhang, L.; Tigabu, B.; Hill, T.E.; Yun, T.; Pietzsch, C.; et al. Favipiravir (T-705) inhibits Junín virus infection and reduces mortality in a guinea pig model of Argentine hemorrhagic fever. PLoS Negl. Trop. Dis. 2013, 7, e2614. [Google Scholar] [CrossRef]
- Herring, S.; Oda, J.M.; Wagoner, J.; Kirchmeier, D.; O’Connor, A.; Nelson, E.A.; Huang, Q.; Liang, Y.; DeWald, L.E.; Johansen, L.M.; et al. Inhibition of Arenaviruses by combinations of orally available approved drugs. Antimicrob. Agents Chemother. 2021, 65, e01146-20. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), A broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, R.A.; Dai, D.; Hosack, V.T.; Tan, Y.; Bolken, T.C.; Hruby, D.E.; Amberg, S.M. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol. 2008, 82, 10768–10775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolken, T.C.; Laquerre, S.; Zhang, Y.; Bailey, T.R.; Pevear, D.C.; Kickner, S.S.; Sperzel, L.E.; Jones, K.F.; Warren, T.K.; Amanda Lund, S.; et al. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antivir. Res. 2006, 69, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Plewe, M.B.; Gantla, V.R.; Sokolova, N.V.; Shin, Y.J.; Naik, S.; Brown, E.R.; Fetsko, A.; Zhang, L.; Kalveram, B.; Freiberg, A.N.; et al. Discovery of a novel highly potent broad-spectrum heterocyclic chemical series of arenavirus cell entry inhibitors. Bioorg. Med. Chem Lett. 2021, 41, 127983. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.M.; Rojek, J.M.; Spiropoulou, C.F.; Gundersen, A.T.; Jin, W.; Shaginian, A.; York, J.; Nunberg, J.H.; Boger, D.L.; Oldstone, M.B.; et al. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem. 2008, 283, 18734–18742. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Bederka, L.H.; Stein, D.A.; Ting, J.P.; Moulton, H.M.; Buchmeier, M.J. Development of peptide-conjugated morpholino oligomers as pan-arenavirus inhibitors. Antimicrob. Agents Chemother. 2011, 55, 4631–4638. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, L.; Cross, R.W.; Geisbert, J.B.; Borisevich, V.; Agans, K.N.; Prasad, A.N.; Enterlein, S.; Aman, M.J.; Bornholdt, Z.A.; Brennan, M.B.; et al. Therapy for Argentine hemorrhagic fever in nonhuman primates with a humanized monoclonal antibody. Proc. Natl. Acad. Sci. USA 2021, 118, e2023332118. [Google Scholar] [CrossRef]
- Pan, X.; Wu, Y.; Wang, W.; Zhang, L.; Xiao, G. Development of horse neutralizing immunoglobulin and immunoglobulin fragments against Junín virus. Antivir. Res. 2020, 174, 104666. [Google Scholar] [CrossRef]
- Linero, F.; Sepúlveda, C.; Christopoulou, I.; Hulpiau, P.; Scolaro, L.; Saelens, X. Neutralization of Junín virus by single domain antibodies targeted against the nucleoprotein. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Pan, X.; Wu, Y.; Wang, W.; Zhang, L.; Xiao, G. Novel neutralizing monoclonal antibodies against Junin virus. Antivir. Res. 2018, 156, 21–28. [Google Scholar] [CrossRef]
- Amanat, F.; Duehr, J.; Oestereich, L.; Hastie, K.M.; Ollmann Saphire, E.; Krammer, F. Antibodies to the Glycoprotein GP2 Subunit Cross-React between Old and New World Arenaviruses. mSphere 2018, 3, e00189-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero, S.; Flores, M.D.; Short, C.; Vazquez, C.A.; Clark, L.E.; Ziegenbein, J.; Zink, S.; Fuentes, D.; Payes, C.; Batto, M.V.; et al. antibody-based Inhibition of pathogenic New World hemorrhagic fever Mammarenaviruses by steric occlusion of the human transferrin receptor 1 apical domain. J. Virol. 2021, 95, e0186820. [Google Scholar] [CrossRef] [PubMed]
- Hickerson, B.T.; Daniels-Wells, T.R.; Payes, C.; Clark, L.E.; Candelaria, P.V.; Bailey, K.W.; Sefing, E.J.; Zink, S.; Ziegenbein, J.; Abraham, J.; et al. Host receptor-targeted therapeutic approach to counter pathogenic New World mammarenavirus infections. Nat. Commun. 2022, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.M.; Sahin, M.; Krumm, S.A.; Seow, J.; Zeltina, A.; Harlos, K.; Paesen, G.C.; Pinschewer, D.D.; Doores, K.J.; Bowden, T.A. Contrasting modes of New World Arenavirus neutralization by immunization-elicited monoclonal antibodies. ASM J. 2022, 22, e0265021. [Google Scholar] [CrossRef]
- Nunberg, J.H.; York, J. The curious case of arenavirus entry, and its inhibition. Viruses 2012, 4, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Burri, D.J.; da Palma, J.R.; Kunz, S.; Pasquato, A. Envelope glycoprotein of arenaviruses. Viruses 2012, 4, 2162–2181. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; de la Torre, J.C.; Peters, C.J. Arenaviridae: The viruses and their replication. In Fields Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Gri, N., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1791–1827. ISBN 9780781760607. [Google Scholar]
- Kiening, M.; Weber, F.; Frishman, D. Conserved RNA structures in the intergenic regions of ambisense viruses. Sci. Rep. 2017, 7, 16625. [Google Scholar] [CrossRef] [Green Version]
- Radoshitzky, S.R.; Buchmeier, M.J.; Charrel, R.N.; Clegg, J.C.S.; Gonzalez, J.J.; Günther, S.; Hepojoki, J.; Kuhn, J.H.; Lukashevich, I.S.; Romanowski, V.; et al. ICTV virus taxonomy profile: Arenaviridae. J. Gen. Virol. 2019, 100, 1200–1201. [Google Scholar] [CrossRef]
- World Health Organization. List of Blueprint Priority Diseases 2022. Available online: https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts (accessed on 25 April 2022).
- York, J.; Agnihothram, S.S.; Romanowski, V.; Nunberg, J.H. Genetic analysis of heptad-repeat regions in the G2 fusion subunit of the Junín arenavirus envelope glycoprotein. Virology 2005, 343, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Agnihothram, S.S.; York, J.; Nunberg, J.H. Role of the stable signal peptide and cytoplasmic domain of G2 in regulating intracellular transport of the Junín virus envelope glycoprotein complex. J. Virol. 2006, 80, 5189–5198. [Google Scholar] [CrossRef] [Green Version]
- York, J.; Nunberg, J.H. Role of the stable signal peptide of Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J. Virol. 2006, 80, 7775–7780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Kwong, J.A.; Albariño, C.G.; Lu, J.G.; Radoshitzky, S.R.; Salazar-Bravo, J.; Farzan, M.; Spiropoulou, C.F.; Choe, H. Host-species transferrin receptor 1 orthologs are cellular receptors for nonpathogenic new world clade B arenaviruses. PLoS Pathog. 2009, 5, e1000358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, A.; Seregin, A.; Huang, C.; Kolokoltsova, O.; Brasier, A.; Peters, C.; Paessler, S. Junín virus pathogenesis and virus replication. Viruses 2012, 4, 2317–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, E. Animal models, prophylaxis, and therapeutics for arenavirus infections. Viruses 2012, 4, 1802–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickerson, B.T.; Westover, J.B.; Wang, Z.; Lee, Y.M.; Gowen, B.B. Guinea pig transferrin receptor 1 mediates cellular entry of Junín Virus and other pathogenic New World arenaviruses. J. Virol. 2020, 94, e01278-e19. [Google Scholar] [CrossRef]
- Martin, V.K.; Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Cannon, P.M. Investigation of clade B New World arenavirus tropism by using chimeric GP1 proteins. J. Virol. 2010, 84, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef] [Green Version]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.G.; Bialecki, M.A.; Belouzard, S.; Cordo, S.M.; Candurra, N.A.; Whittaker, G.R. Utilization of human DC-SIGN and L-SIGN for entry and infection of host cells by the New World arenavirus, Junín virus. Biochem. Biophys Res. Commun. 2013, 441, 612–617. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillette, R.B.; Phillips, E.K.; Patel, R.; Mahauad-Fernandez, W.; Moller-Tank, S.; Rogers, K.J.; Dillard, J.A.; Cooney, A.L.; Martinez-Sobrido, L.; Okeoma, C.; et al. TIM-1 mediates dystroglycan-independent entry of Lassa virus. J. Virol. 2018, 92, e00093-e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohan, D.; Van Ert, H.; Ruggio, N.; Rogers, K.J.; Badreddine, M.; Aguilar Briseño, J.A.; Elliff, J.M.; Rojas Chavez, R.A.; Gao, B.; Stokowy, T.; et al. Phosphatidylserine receptors enhance SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009743. [Google Scholar] [CrossRef] [PubMed]
- Svajger, U.; Anderluh, M.; Jeras, M.; Obermajer, N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010, 22, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Gramberg, T.; Soilleux, E.; Fisch, T.; Lalor, P.F.; Hofmann, H.; Wheeldon, S.; Cotterill, A.; Wegele, A.; Winkler, T.; Adams, D.H.; et al. Interactions of LSECtin and DC- SIGN/DC-SIGNR with viral ligands: Differential pH dependence, internalization and virion binding. Virology 2008, 373, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Sarute, N.; Ross, S.R. CACNA1S haploinsufficiency confers resistance to New World arenavirus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 19497–19506. [Google Scholar] [CrossRef]
- Pelkmans, L.; Helenius, A. Insider information: What viruses tell us about endocytosis. Curr. Opin. Cell Biol. 2003, 15, 414–422. [Google Scholar] [CrossRef]
- Ripa, I.; Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Membrane Rafts: Portals for Viral Entry. Front. Microbiol. 2021, 12, 631274. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Characterization of Junin arenavirus cell entry. J. Gen. Virol. 2007, 88, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Forlenza, M.B.; Candurra, N.A. Involvement of cellular proteins in Junin arenavirus entry. Biotechnol. J. 2009, 4, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Involvement of cytoskeleton in Junín virus entry. Virus Res. 2008, 138, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Yarar, D.; Waterman-Storer, C.M.; Schmid, S.L. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol. Biol. Cell 2005, 16, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Castilla, V.; Mersich, S.E.; Candurra, N.A.; Damonte, E.B. The entry of Junin virus into Vero cells. Arch. Virol. 1994, 136, 363–374. [Google Scholar] [CrossRef]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Castilla, V.; Palermo, L.M.; Coto, C.E. Involvement of vacuolar proton ATPase in Junin virus multiplication. Arch. Virol. 2001, 146, 251–263. [Google Scholar] [CrossRef]
- Borchers, A.C.; Langemeyer, L.; Ungermann, C. Who’s in control? Principles of Rab GTPase activation in endolysosomal membrane trafficking and beyond. J. Cell Biol. 2021, 220, e202105120. [Google Scholar] [CrossRef]
- Eschli, B.; Quirin, K.; Wepf, A.; Weber, J.; Zinkernagel, R.; Hengartner, H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J. Virol. 2006, 80, 5897–5907. [Google Scholar] [CrossRef] [Green Version]
- Di Simone, C.; Zandonatti, M.A.; Buchmeier, M.J. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology 1994, 198, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Klewitz, C.; Klenk, H.D.; Ter Meulen, J. Amino acids from both N-terminal hydrophobic regions of the Lassa virus envelope glycoprotein GP-2 are critical for pH-dependent membrane fusion and infectivity. J. Gen. Virol. 2007, 88, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Igonet, S.; Vaney, M.C.; Vonrhein, C.; Bricogne, G.; Stura, E.A.; Hengartner, H.; Eschli, B.; Rey, F.A. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc. Natl. Acad. Sci. USA 2011, 108, 19967–19972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M. TRIM proteins: Another class of viral victims. Sci. Signal. 2010, 3, jc2. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef]
- Rajsbaum, R.; García-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [Green Version]
- Sarute, N.; Ibrahim, N.; Medegan Fagla, B.; Lavanya, M.; Cuevas, C.; Stavrou, S.; Otkiran-Clare, G.; Tyynismaa, H.; Henao-Mejia, J.; Ross, S.R. TRIM2, a novel member of the antiviral family, limits New World arenavirus entry. PLoS Biol. 2019, 17, e3000137. [Google Scholar] [CrossRef]
- Sarute, N.; Cheng, H.; Yan, Z.; Salas-Briceno, K.; Richner, J.; Rong, L.; Ross, S.R. Signal-regulatory protein alpha is an anti-viral entry factor targeting viruses using endocytic pathways. PLoS Pathog. 2021, 17, e1009662. [Google Scholar] [CrossRef]
- Pyle, J.D.; Whelan, S.P.J. Isolation of reconstructed functional ribonucleoprotein complexes of Machupo virus. J. Virol. 2021, 95, e0105421. [Google Scholar] [CrossRef]
- López, N.; Jácamo, R.; Franze-Fernández, M.T. Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J. Virol. 2001, 75, 12241–12251. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Novella, I.S.; Teng, M.N.; Oldstone, M.B.; de La Torre, J.C. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 2000, 74, 3470–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, M.; Gölnitz, U.; Müller, S.; Becker-Ziaja, B.; Günther, S. Replicon system for Lassa virus. J. Virol. 2004, 78, 13793–13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornu, T.I.; de la Torre, J.C. RING finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J. Virol. 2001, 75, 9415–9426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jácamo, R.; López, N.; Wilda, M.; Franze-Fernández, M.T. Tacaribe virus Z protein interacts with the L polymerase protein to inhibit viral RNA synthesis. J. Virol. 2003, 77, 10383–10393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranzusch, P.J.; Whelan, S.P. Arenavirus Z protein controls viral RNA synthesis by locking a polymerase-promoter complex. Proc. Natl. Acad. Sci. USA 2011, 108, 19743–19748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, M.E.; Wilda, M.; Levingston Macleod, J.M.; D’Antuono, A.; Foscaldi, S.; Marino Buslje, C.; Lopez, N. Molecular determinants of arenavirus Z protein homo-oligomerization and L polymerase binding. J. Virol. 2011, 85, 12304–12314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Cong, J.; Wang, C.; Ji, W.; Xin, Y.; Qian, Y.; Li, X.; Chen, Y.; Rao, Z. Structural basis for recognition and regulation of arenavirus polymerase L by Z protein. Nat. Commun. 2021, 12, 4134. [Google Scholar] [CrossRef]
- Eichler, R.; Strecker, T.; Kolesnikova, L.; ter Meulen, J.; Weissenhorn, W.; Becker, S.; Klenk, H.D.; Garten, W.; Lenz, O. Characterization of the Lassa virus matrix protein Z: Electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 2004, 100, 249–255. [Google Scholar] [CrossRef]
- Casabona, J.C.; Levingston Macleod, J.M.; Loureiro, M.E.; Gomez, G.A.; Lopez, N. The RING domain and the L79 residue of Z protein are involved in both the rescue of nucleocapsids and the incorporation of glycoproteins into infectious chimeric arenavirus-like particles. J. Virol. 2009, 83, 7029–7039. [Google Scholar] [CrossRef] [Green Version]
- Capul, A.A.; Perez, M.; Burke, E.; Kunz, S.; Buchmeier, M.J.; de la Torre, J.C. Arenavirus Z-glycoprotein association requires Z myristoylation but not functional RING or late domains. J. Virol. 2007, 81, 9451–9460. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strecker, T.; Eichler, R.; Meulen, J.T.; Weissenhorn, W.; Dieter Klenk, H.; Garten, W.; Lenz, O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groseth, A.; Wolff, S.; Strecker, T.; Hoenen, T.; Becker, S. Efficient budding of the tacaribe virus matrix protein z requires the nucleoprotein. J. Virol. 2010, 84, 3603–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef] [Green Version]
- King, B.R.; Kellner, S.; Eisenhauer, P.L.; Bruce, E.A.; Ziegler, C.M.; Zenklusen, D.; Botten, J.W. Visualization of the lymphocytic choriomeningitis mammarenavirus (LCMV) genome reveals the early endosome as a possible site for genome replication and viral particle pre-assembly. J. Gen. Virol. 2017, 98, 2454–2460. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. AMS J. 2018, 9, e02290-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles Virus Forms Inclusion Bodies with Properties of Liquid Organelles. J. Virol. 2019, 93, e00948-19. [Google Scholar] [CrossRef]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentín, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef]
- Tourrière, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef]
- King, B.R.; Hershkowitz, D.; Eisenhauer, P.L.; Weir, M.E.; Ziegler, C.M.; Russo, J.; Bruce, E.A.; Ballif, B.A.; Botten, J. A map of the Arenavirus Nucleoprotein-Host protein interactome reveals that Junín virus selectively impairs the antiviral activity of double-stranded RNA-activated Protein Kinase (PKR). J. Virol. 2017, 91, e00763-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linero, F.N.; Thomas, M.G.; Boccaccio, G.L.; Scolaro, L.A. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2α phosphorylation. J. Gen. Virol. 2011, 92, 2889–2899. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, M.E.; D’Antuono, A.; Levingston Macleod, J.M.; López, N. Uncovering viral protein-protein interactions and their role in arenavirus life cycle. Viruses 2012, 4, 1651–1667. [Google Scholar] [CrossRef]
- Fehling, S.K.; Lennartz, F.; Strecker, T. Multifunctional nature of the arenavirus RING finger protein, Z. Viruses 2012, 4, 2973–3011. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, C.M.; Eisenhauer, P.; Kelly, J.A.; Dang, L.N.; Beganovic, V.; Bruce, E.A.; King, B.R.; Shirley, D.J.; Weir, M.E.; Ballif, B.A.; et al. A Proteomics survey of Junín virus interactions with human proteins reveals host factors required for arenavirus replication. J. Virol. 2018, 92, e01565-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, M.E.; Zorzetto-Fernandes, A.L.; Radoshitzky, S.; Chi, X.; Dallari, S.; Marooki, N.; Lèger, P.; Foscaldi, S.; Harjono, V.; Sharma, S.; et al. DDX3 suppresses type I interferons and favors viral replication during Arenavirus infection. PLoS Pathog. 2018, 14, e1007125. [Google Scholar] [CrossRef]
- Ariumi, Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014, 5, 423. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Díaz, T.; Valiente-Echeverría, F.; Soto-Rifo, R. RNA Helicase DDX3: A Double-Edged Sword for Viral Replication and Immune Signaling. Microorganisms 2021, 9, 1206. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Abe, K.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato, N. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 2007, 81, 13922–13926. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Wang, S.W.; Cheng, L.; Tarn, W.Y.; Tsai, S.J.; Sun, H.S. Human DDX3 interacts with the HIV-1 Tat protein to facilitate viral mRNA translation. PLoS ONE 2013, 8, e68665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Rifo, R.; Rubilar, P.S.; Limousin, T.; de Breyne, S.; Décimo, D.; Ohlmann, T. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 2012, 31, 3745–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- DuShane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar] [CrossRef] [Green Version]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Yang, S.H.; Sharrocks, A.D.; Whitmarsh, A.J. MAP kinase signalling cascades and transcriptional regulation. Gene 2013, 513, 1–13. [Google Scholar] [CrossRef]
- Plotnikov, E.; Zehorai, S.; Procaccia, R. Seger The MAPK cascades: Signalling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.E.; Brunetti, J.E.; Wachsman, M.B.; Scolaro, L.A.; Castilla, V. Raf/MEK/ERK pathway activation is required for Junín virus replication. J. Gen. Virol. 2014, 95, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, J.E.; Foscaldi, S.; Quintana, V.M.; Scolaro, L.A.; López, N.; Castilla, V. Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses. Viruses 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Fuentes, S.M.; Timani, K.; Sun, D.; Murphy, C.; Lin, Y.; August, A.; Teng, M.N.; He, B. Akt plays a critical role in replication of nonsegmented negative-stranded RNA viruses. J. Virol. 2008, 82, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, S.; Ngo, N.; de la Torre, J.C. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 2012, 86, 4578–4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linero, F.N.; Scolaro, L.A. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junín virus replication in vitro. Virus Res. 2009, 145, 166–170. [Google Scholar] [CrossRef]
- Hedstrom, L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Smith, J.; Bukreyeva, N.; Koma, T.; Manning, J.T.; Kalkeri, R.; Kwong, A.D.; Paessler, S. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antivir. Res. 2018, 149, 34–40. [Google Scholar] [CrossRef]
- Dunham, E.C.; Leske, A.; Shifflett, K.; Watt, A.; Feldmann, H.; Hoenen, T.; Groseth, A. Lifecycle modelling systems support inosine monophosphate dehydrogenase (IMPDH) as a pro-viral factor and antiviral target for New World arenaviruses. Antivir. Res. 2018, 157, 140–150. [Google Scholar] [CrossRef]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Thibault, P.A.; Ganesan, A.; Kalyaanamoorthy, S.; Clarke, J.W.E.; Salapa, H.E.; Levin, M.C. hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease. Biology 2021, 10, 712. [Google Scholar] [CrossRef]
- Chang, C.K.; Chen, C.J.; Wu, C.C.; Chen, S.W.; Shih, S.R.; Kuo, R.L. Cellular hnRNP A2/B1 interacts with the NP of influenza A virus and impacts viral replication. PLoS ONE 2017, 12, e0188214. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, K.; Halvorsen, M.; Abrahamyan, L.; Chatel-Chaix, L.; Poupon, V.; Gordon, H.; Des Groseillers, L.; Gatignol, A.; Mouland, A.J. Trafficking of HIV-1 RNA is mediated by heterogeneous nuclear ribonucleoprotein A2 expression and impacts on viral assembly. Traffic 2006, 7, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Dinh, P.X.; Das, A.; Franco, R.; Pattnaik, A.K. Heterogeneous nuclear ribonucleoprotein K supports vesicular stomatitis virus replication by regulating cell survival and cellular gene expression. J. Virol. 2013, 87, 10059–10069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeto, C.A.; Knott, M.E.; Linero, F.N.; Ellenberg, P.C.; Scolaro, L.A.; Castilla, V. Differential effect of acute and persistent Junin virus infections on the nucleo-cytoplasmic trafficking and expression of heterogeneous nuclear ribonucleoproteins type, A. and, B. J. Gen. Virol. 2011, 92, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, J.E.; Scolaro, L.A.; Castilla, V. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) is a host factor required for dengue virus and Junín virus multiplication. Virus Res. 2015, 203, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kelen, K.; Beyaert, R.; Inzé, D.; De Veylder, L. Translational control of eukaryotic gene expression. Crit Rev. Biochem Mol. Biol. 2009, 44, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Au, H.H.; Jan, E. Novel viral translation strategies. Wiley Interdiscip Rev. RNA. 2014, 5, 779–801. [Google Scholar] [CrossRef]
- Jan, E.; Mohr, I.; Walsh, D. A Cap-to-Tail Guide to mRNA Translation Strategies in Virus-Infected Cells. Annu. Rev. Virol. 2016, 3, 283–307. [Google Scholar] [CrossRef]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.C. HIV-1 replication and the cellular eukaryotic translation apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [Green Version]
- Yángüez, E.; Rodriguez, P.; Goodfellow, I.; Nieto, A. Influenza virus polymerase confers independence of the cellular cap-binding factor eIF4E for viral mRNA translation. Virology 2012, 422, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.A.; Panganiban, A.T. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008, 27, 3129–3139. [Google Scholar] [CrossRef] [Green Version]
- de Breyne, S.; Chamond, N.; Décimo, D.; Trabaud, M.A.; André, P.; Sargueil, B.; Ohlmann, T. In vitro studies reveal that different modes of initiation on HIV-1 mRNA have different levels of requirement for eukaryotic initiation factor 4F. FEBS J. 2012, 279, 3098–3111. [Google Scholar] [CrossRef] [PubMed]
- Linero, F.; Welnowska, E.; Carrasco, L.; Scolaro, L. Participation of eIF4F complex in Junin virus infection: Blockage of eIF4E does not impair virus replication. Cell Microbiol. 2013, 15, 1766–1782. [Google Scholar] [CrossRef] [PubMed]
- Foscaldi, S.; D′Antuono, A.; Noval, M.G.; de Prat Gay, G.; Scolaro, L.; Lopez, N. Regulation of Tacaribe Mammarenavirus Translation: Positive 5′ and Negative 3′ Elements and Role of Key Cellular Factors. J. Virol. 2017, 91, e00084-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell-Dwyer, E.J.; Lai, H.; MacDonald, R.C.; Salvato, M.S.; Borden, K.L. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J. Virol. 2000, 74, 3293–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kentsis, A.; Dwyer, E.C.; Perez, J.M.; Sharma, M.; Chen, A.; Pan, Z.Q.; Borden, K.L. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J. Mol. Biol. 2001, 312, 609–623. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc. Natl. Acad. Sci. USA 2010, 107, 5441–5446. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, A.M.; Labonté, P. The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci. Rep. 2017, 7, 40351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011, 85, 6319–6333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Baillet, N.; Krieger, S.; Journeaux, A.; Caro, V.; Tangy, F.; Vidalain, P.O.; Baize, S. Autophagy promotes infectious particle production of Mopeia and Lassa viruses. Viruses 2019, 11, 293. [Google Scholar] [CrossRef] [Green Version]
- Roldán, J.S.; Candurra, N.A.; Colombo, M.I.; Delgui, L.R. Junín virus promotes autophagy to facilitate the virus life cycle. J. Virol. 2019, 93, e02307-18. [Google Scholar] [CrossRef] [Green Version]
- Perez Vidakovics, M.L.A.; Ure, A.E.; Arrías, P.N.; Romanowski, V.; Gómez, R.M. Junín virus induces autophagy in human A549 cells. PLoS ONE 2019, 14, e0218730. [Google Scholar] [CrossRef]
- Gallo, G.L.; Roldán, J.S.; Delgui, L.R. Autophagy modulation in Mammarenavirus infection. J. Immunol. Sci. 2020, 4, 45–51. [Google Scholar] [CrossRef]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Lippens, S.; Vanden Berghe, T.; Romagnoli, A.; Fimia, G.M.; Piacentini, M.; Vandenabeele, P. Beclin1: A role in membrane dynamics and beyond. Autophagy 2012, 8, 6–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliari, F.; Marafioti, M.G.; Genard, G.; Candeloro, P.; Viglietto, G.; Seco, J.; Tirinato, L. ssRNA Virus and Host Lipid Rearrangements: Is There a Role for Lipid Droplets in SARS-CoV-2 Infection? Front. Mol. Biosci. 2020, 7, 578964. [Google Scholar] [CrossRef]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assunção-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal Structure of the Capsid Protein from Zika Virus. J. Mol. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef]
- Bang, B.R.; Li, M.; Tsai, K.N.; Aoyagi, H.; Lee, S.A.; Machida, K.; Aizaki, H.; Jung, J.U.; Ou, J.J.; Saito, T. Regulation of Hepatitis C Virus Infection by Cellular Retinoic Acid Binding Proteins through the Modulation of Lipid Droplet Abundance. J. Virol. 2019, 93, e02302-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010, 8, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018, 23, 819–831.e5. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Dias, S.S.; Cardoso Soares, V.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef]
- Peña Cárcamo, J.R.; Morell, M.L.; Vázquez, C.A.; Vatansever, S.; Upadhyay, A.S.; Överby, A.K.; Cordo, S.M.; García, C.C. The interplay between viperin antiviral activity, lipid droplets and Junín mammarenavirus multiplication. Virology 2018, 514, 216–229. [Google Scholar] [CrossRef]
- Cordo, S.M.; Candurra, N.A.; Damonte, E.B. Myristic acid analogs are inhibitors of Junin virus replication. Microbes Infect. 1999, 1, 609–614. [Google Scholar] [CrossRef]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Wieland, F.; Harter, C. Mechanisms of vesicle formation: Insights from the COP system. Curr. Opin. Cell Biol. 1999, 11, 440–446. [Google Scholar] [CrossRef]
- Martínez, J.L.; Arias, C.F. Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses. Viruses 2020, 12, 682. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, J.T.; Yun, N.E.; Seregin, A.V.; Koma, T.; Sattler, R.A.; Ezeomah, C.; Huang, C.; de la Torre, J.C.; Paessler, S. The glycoprotein of the live-attenuated Junin virus vaccine strain induces endoplasmic reticulum stress and forms aggregates prior to degradation in the lysosome. J. Virol. 2020, 94, e01693-e19. [Google Scholar] [CrossRef] [PubMed]
- Klaus, J.P.; Eisenhauer, P.; Russo, J.; Mason, A.B.; Do, D.; King, B.; Taatjes, D.; Cornillez-Ty, C.; Boyson, J.E.; Thali, M.; et al. The intracellular cargo receptor ERGIC-53 is required for the production of infectious arenavirus, coronavirus, and filovirus particles. Cell Host Microbe. 2013, 14, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Cordo, S.M.; Valko, A.; Martinez, G.M.; Candurra, N.A. Membrane localization of Junín virus glycoproteins requires cholesterol and cholesterol rich membranes. Biochem. Biophys. Res. Commun. 2013, 430, 912–917. [Google Scholar] [CrossRef]
- Agnihothram, S.S.; Dancho, B.; Grant, K.W.; Grimes, M.L.; Lyles, D.S.; Nunberg, J.H. Assembly of arenavirus envelope glycoprotein GPC in detergent-soluble membrane microdomains. J. Virol. 2009, 83, 9890–9900. [Google Scholar] [CrossRef] [Green Version]
- Gaudin, R.; Barteneva, N.S. Sorting of small infectious virus particles by flow virometry reveals distinct infectivity profiles. Nat. Commun. 2015, 6, 6022. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for Lassa virus budding. J. Virol. 2006, 80, 4191–4195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welker, L.; Paillart, J.C.; Bernacchi, S. Importance of viral late domains in budding and release of enveloped RNA viruses. Viruses 2021, 13, 1559. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J. Interferon. Res. 1985, 5, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Marta, R.F.; Montero, V.S.; Hack, C.E.; Sturk, A.; Maiztegui, J.I.; Molinas, F.C. Proinflammatory cytokines and elastase-alpha-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 1999, 60, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, M.V.; Saavedra, M.C.; Falcoff, R.; Maiztegui, J.I.; Molinas, F.C. Increased tumor necrosis factor-alpha levels in Argentine hemorrhagic fever. J. Infect. Dis. 1992, 166, 1203–1204. [Google Scholar] [CrossRef]
- Mantlo, E.; Paessler, S.; Huang, C. Differential immune responses to hemorrhagic fever-causing arenaviruses. Vaccines 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Rojas, J.M.; Alejo, A.; Martín, V.; Sevilla, N. Viral pathogen-induced mechanisms to antagonize mammalian interferon (IFN) signaling pathway. Cell Mol. Life Sci. 2021, 78, 1423–1444. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; de la Torre, J.C.; et al. Junín virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateer, E.J.; Paessler, S.; Huang, C. Visualization of double-stranded RNA colocalizing with pattern recognition receptors in arenavirus infected cells. Front. Cell Infect. Microbiol. 2018, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, H.; Möller, R.; Fedeli, C.; Gerold, G.; Kunz, S. Comparison of the innate immune responses to pathogenic and nonpathogenic Clade B New World arenaviruses. J. Virol. 2019, 93, e00148-e19. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Ronca, S.; Koma, T.; Paessler, S. Highly pathogenic New World and Old World human arenaviruses induce distinct interferon responses in human cells. J. Virol. 2015, 89, 7079–7088. [Google Scholar] [CrossRef] [Green Version]
- Negrotto, S.; Mena, H.A.; Ure, A.E.; Jaquenod De Giusti, C.; Bollati-Fogolín, M.; Vermeulen, E.M.; Schattner, M.; Gómez, R.M. Human plasmacytoid dendritic cells elicited different responses after infection with pathogenic and nonpathogenic Junin virus strains. J. Virol. 2015, 89, 7409–7413. [Google Scholar] [CrossRef] [Green Version]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Mateer, E.J.; Koma, T.; Paessler, S. Highly Pathogenic New World Arenavirus infection activates the pattern recognition receptor Protein Kinase R without attenuating virus replication in human cells. J. Virol. 2017, 91, e01090-17. [Google Scholar] [CrossRef] [Green Version]
- Mateer, E.J.; Maruyama, J.; Card, G.E.; Paessler, S.; Huang, C. Lassa virus, but not highly pathogenic New World arenaviruses, restricts immunostimulatory double-stranded RNA accumulation during Infection. J. Virol. 2020, 94, e02006-19. [Google Scholar] [CrossRef]
- Moreno, H.; Kunz, S. The Protein Kinase Receptor modulates the innate immune response against Tacaribe virus. Viruses 2021, 13, 1313. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Ross, S.R. Toll-like receptor 2-mediated innate immune responses against Junín virus in mice lead to antiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinson, E.R.; Cresswell, P. The antiviral protein viperin localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc. Natl. Acad. Sci. USA 2009, 106, 20452–20457. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, V.R.; Urata, S.; Sakaguchi, M.; Yasuda, J. Human BST-2/tetherin inhibits Junin virus release from host cells and its inhibition is partially counteracted by viral nucleoprotein. J. Gen. Virol. 2020, 101, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; de la Torre, J.C.; McGavern, D.B.; Nayak, D. Beyond Tethering the Viral Particles: Immunomodulatory Functions of Tetherin (BST-2). DNA Cell Biol. 2019, 38, 1170–1177. [Google Scholar] [CrossRef] [Green Version]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Moreno, R.; Martínez-Romero, C.; García-Sastre, A. Induction and Evasion of Type-I Interferon Responses during Influenza A Virus Infection. Cold Spring Harb. Perspect. Med. 2021, 11, a038414. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O′Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef]
- Martínez-Sobrido, L.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa, B. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.; Huang, Q.; Liu, X.; Di, D.; Liang, Y.; Ly, H. Arenaviral Nucleoproteins Suppress PACT-Induced Augmentation of RIG-I Function To Inhibit Type I Interferon Production. J. Virol. 2018, 92, e00482-e18. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; de la Torre, J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef] [Green Version]
- Reynard, S.; Russier, M.; Fizet, A.; Carnec, X.; Baize, S. Exonuclease domain of the Lassa virus nucleoprotein is critical to avoid RIG-I signaling and to inhibit the innate immune response. J. Virol. 2014, 88, 13923–13927. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3’ to 5’ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef] [Green Version]
- Hastie, K.M.; Liu, T.; Li, S.; King, L.B.; Ngo, N.; Zandonatti, M.A.; Woods, V.L., Jr.; de la Torre, J.C.; Saphire, E.O. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. USA 2011, 108, 19365–19370. [Google Scholar] [CrossRef] [Green Version]
- Harmon, B.; Kozina, C.; Maar, D.; Carpenter, T.S.; Branda, C.S.; Negrete, O.A.; Carson, B.D. Identification of critical amino acids within the nucleoprotein of Tacaribe virus important for anti-interferon activity. J. Biol. Chem. 2013, 288, 8702–8711. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Shao, J.; Lan, S.; Zhou, Y.; Xing, J.; Dong, C.; Liang, Y.; Ly, H. In vitro and in vivo characterizations of pichinde viral nucleoprotein exoribonuclease functions. J. Virol. 2015, 89, 6595–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnec, X.; Mateo, M.; Page, A.; Reynard, S.; Hortion, J.; Picard, C.; Yekwa, E.; Barrot, L.; Barron, S.; Vallve, A.; et al. A Vaccine platform against arenaviruses based on a recombinant hyperattenuated Mopeia virus expressing heterologous glycoproteins. J. Virol. 2018, 92, e02230-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, L.; Liu, X.; Dong, S.; Wang, W.; Huo, T.; Guo, Y.; Rao, Z.; Yang, C. Crystal structure of Junin virus nucleoprotein. J. Gen. Virol. 2013, 94, 2175–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Chai, Z.; Ly, H.; Liang, Y. Differential Inhibition of Macrophage Activation by Lymphocytic Choriomeningitis Virus and Pichinde Virus Is Mediated by the Z Protein N-Terminal Domain. J. Virol. 2015, 89, 12513–12517. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Ly, H.; Liang, Y. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-I-like receptor-dependent interferon production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [Green Version]
- Albariño, C.G.; Bergeron, E.; Erickson, B.R.; Khristova, M.L.; Rollin, P.E.; Nichol, S.T. Efficient reverse genetics generation of infectious junin viruses differing in glycoprotein processing. J. Virol. 2009, 83, 5606–5614. [Google Scholar] [CrossRef] [Green Version]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; White, D.M.; Bergeron, E.; Shrivastava-Ranjan, P.; Nichol, S.T. Reverse genetics generation of chimeric infectious Junin/Lassa virus is dependent on interaction of homologous glycoprotein stable signal peptide and G2 cytoplasmic domains. J. Virol. 2011, 85, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Emonet, S.F.; Seregin, A.V.; Yun, N.E.; Poussard, A.L.; Walker, A.G.; de la Torre, J.C.; Paessler, S. Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid#1 strains of Junin virus, the causative agent of Argentine hemorrhagic fever disease. J. Virol. 2011, 85, 1473–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Riaño, E.; Cheng, B.Y.H.; Carlos de la Torre, J.; Martínez-Sobrido, L. Arenavirus reverse genetics for vaccine development. J. Gen. Virol. 2013, 94, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Seregin, A.V.; Yun, N.E.; Miller, M.; Aronson, J.; Smith, J.K.; Walker, A.G.; Smith, J.N.; Huang, C.; Manning, J.T.; de la Torre, J.C.; et al. The glycoprotein precursor gene of Junin virus determines the virulence of the Romero strain and the attenuation of the Candid #1 strain in a representative animal model of Argentine hemorrhagic fever. J. Virol. 2015, 89, 5949–5956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Flint, M.; Bergeron, E.; White, D.M.; Nichol, S.T. The major determinant of attenuation in mice of the Candid1 vaccine for Argentine hemorrhagic fever is located in the G2 glycoprotein transmembrane domain. J. Virol. 2011, 85, 10404–10408. [Google Scholar] [CrossRef] [Green Version]
- Gowen, B.B.; Hickerson, B.T.; York, J.; Westover, J.B.; Sefing, E.J.; Bailey, K.W.; Wandersee, L.; Nunberg, J.H. Second-Generation Live-Attenuated Candid#1 Vaccine Virus Resists Reversion and Protects against Lethal Junín Virus Infection in Guinea Pigs. J. Virol. 2021, 95, e0039721. [Google Scholar] [CrossRef]
- Koma, T.; Huang, C.; Coscia, A.; Hallam, S.; Manning, J.T.; Maruyama, J.; Walker, A.G.; Miller, M.; Smith, J.N.; Patterson, M.; et al. Glycoprotein N-linked glycans play a critical role in arenavirus pathogenicity. PLoS Pathog. 2021, 17, e1009356. [Google Scholar] [CrossRef]
- Hallam, S.J.; Manning, J.T.; Maruyama, J.; Seregin, A.; Huang, C.; Walker, D.H.; de la Torre, J.C.; Paessler, S. A single mutation (V64G) within the RING Domain of Z attenuates Junin virus. PLoS Negl. Trop. Dis. 2020, 14, e0008555. [Google Scholar] [CrossRef]
- Ye, C.; de la Torre, J.C.; Martínez-Sobrido, L. Development of Reverse Genetics for the Prototype New World Mammarenavirus Tacaribe Virus. J. Virol. 2020, 94, e01014–e01020. [Google Scholar] [CrossRef]
- Foscaldi, S.; Loureiro, M.E.; Sepúlveda, C.; Palacios, C.; Forlenza, M.B.; López, N. Development of a Reverse Genetic System to Generate Recombinant Chimeric Tacaribe Virus that Expresses Junín Virus Glycoproteins. Pathogens. 2020, 9, 948. [Google Scholar] [CrossRef]
- Wendt, L.; Bostedt, L.; Hoenen, T.; Groseth, A. High-throughput screening for negative-stranded hemorrhagic fever viruses using reverse genetics. Antivir. Res. 2019, 170, 104569. [Google Scholar] [CrossRef]
- Mpingabo, P.I.; Urata, S.; Yasuda, J. Analysis of the Cell Type-Dependence on the Arenavirus Z-Mediated Virus-Like Particle Production. Front. Microbiol. 2020, 11, 562814. [Google Scholar] [CrossRef] [PubMed]
- Raabe, V.N.; Kann, G.; Ribner, B.S.; Morales, A.; Varkey, J.B.; Mehta, A.K.; Lyon, G.M.; Vanairsdale, S.; Faber, K.; Becker, S.; et al. Favipiravir and ribavirin treatment of epidemiologically linked cases of Lassa Fever. Clin. Infect. Dis. 2017, 65, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veliziotis, I.; Roman, A.; Martiny, D.; Schuldt, G.; Claus, M.; Dauby, N.; Van den Wijngaert, S.; Martin, C.; Nasreddine, R.; Perandones, C.; et al. Clinical management of Argentine Hemorrhagic Fever using ribavirin and favipiravir, Belgium, 2020. Emerg. Infect. Dis. 2020, 26, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Ramírez, G.; Lorenzo, C.; Navarrete, D.; Carrillo-Reyes, A.; Retana, Ó.; Carrasco-Hernández, R. A Review of mammarenaviruses and rodent reservoirs in the Americas. Ecohealth 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Antezana, J.P.; Rodriguez-Villena, O.J.; Arancibia-Alba, A.W.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Rodríguez-Morales, A.J. Clinical features of fatal cases of Chapare virus hemorrhagic fever originating from rural La Paz, Bolivia, 2019: A cluster analysis. Travel Med. Infect. Dis. 2020, 36, 101589. [Google Scholar] [CrossRef]
- de Mello Malta, F.; Amgarten, D.; Nastri, A.C.S.S.; Ho, Y.L.; Boas Casadio, L.V.; Basqueira, M.; Selegatto, G.; Cervato, M.C.; Duarte-Neto, A.N.; Higashino, H.R.; et al. Sabiá virus-like mammarenavirus in patient with fatal hemorrhagic fever, Brazil, 2020. Emerg. Infect. Dis. 2020, 26, 1332–1334. [Google Scholar] [CrossRef]
- Fernandes, J.; Guterres, A.; de Oliveira, R.C.; Jardim, R.; Dávila, A.M.R.; Hewson, R.; de Lemos, E.R.S. Aporé virus, a novel mammarenavirus (Bunyavirales: Arenaviridae) related to highly pathogenic virus from South America. Mem Inst. Oswaldo Cruz. 2019, 114, e180586. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, G.L.; López, N.; Loureiro, M.E. The Virus–Host Interplay in Junín Mammarenavirus Infection. Viruses 2022, 14, 1134. https://doi.org/10.3390/v14061134
Gallo GL, López N, Loureiro ME. The Virus–Host Interplay in Junín Mammarenavirus Infection. Viruses. 2022; 14(6):1134. https://doi.org/10.3390/v14061134
Chicago/Turabian StyleGallo, Giovanna Lucrecia, Nora López, and María Eugenia Loureiro. 2022. "The Virus–Host Interplay in Junín Mammarenavirus Infection" Viruses 14, no. 6: 1134. https://doi.org/10.3390/v14061134
APA StyleGallo, G. L., López, N., & Loureiro, M. E. (2022). The Virus–Host Interplay in Junín Mammarenavirus Infection. Viruses, 14(6), 1134. https://doi.org/10.3390/v14061134