
Description of a One-Year Succession of Variants of Interest and Concern of SARS-CoV-2 in Venezuela

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of VOCs or VOIs by Partial Genome Sequencing
2.2. Complete Genome Sequencing of Selected Isolates
2.3. Statistical Analysis
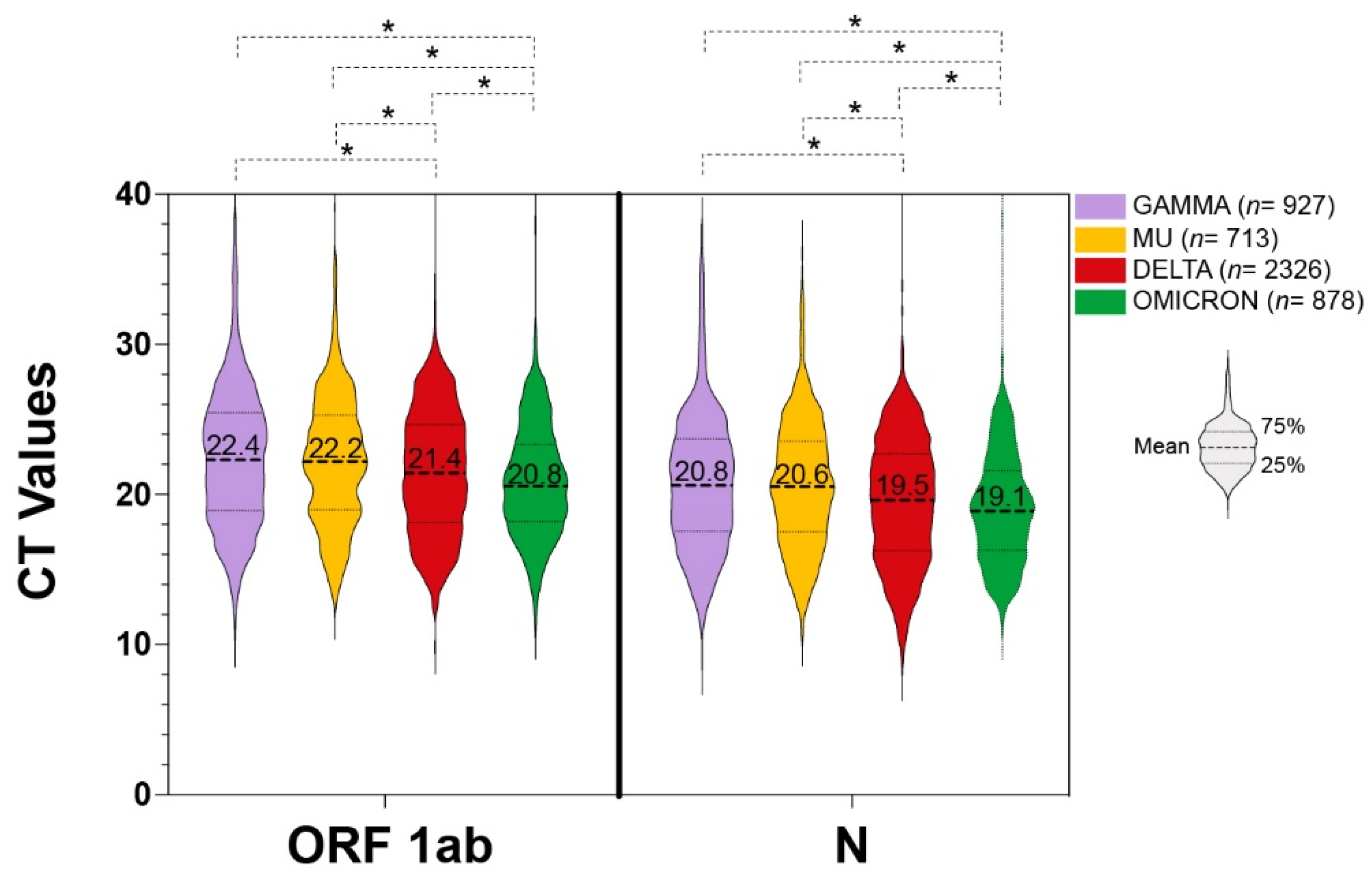
3. Results

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Zhou, J.; Dong, Y.; Jiang, W.; Jiang, W. Rampant C-to-U deamination accounts for the intrinsically high mutation rate in SARS-CoV-2 spike gene. RNA 2022, 28, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Lineage List. Available online: https://cov-lineages.org/lineage_list.html (accessed on 14 May 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 14 May 2022).
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: Matched cohort study. Br. Med. J. 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARSCoV-2 Omicron variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Saunders, K.; Edwards, R.J.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.E.; Dávila-Barclay, A.; Salvatierra, G.; González, L.; Cuicapuza, D.; Solís, L.; Marcos-Carbajal, P.; Huancachoque, J.; Maturrano, L.; Tsukayama, P. The Emergence of Sars-CoV-2 Variant Lambda (C.37) in South America. Microbiol. Spectr. 2021, 9, e0078921. [Google Scholar] [CrossRef] [PubMed]
- Laiton-Donato, K.; Franco-Muñoz, C.; Álvarez-Díaz, D.A.; Ruiz-Moreno, H.A.; Usme-Ciro, J.A.; Prada, D.A.; Reales-González, J.; Corchuelo, S.; Herrera-Sepúlveda, M.T.; Naizaque, J.; et al. Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2. Infect. Genet. Evol. 2021, 95, 105038. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Roustan, C.; Borg, A.; Hussain, S.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Evolution of the SARS-CoV-2 spike protein in the human host. Evolution of the SARS-CoV-2 spike protein in the human host. Nat. Commun. 2022, 13, 1178. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J.; Beale, R. Immunity to SARS-CoV-2 variants of concern. Science 2021, 371, 1103–1104. [Google Scholar] [CrossRef]
- Ferrareze, P.A.G.; Franceschi, V.B.; Mayer, A.M.; Caldana, G.D.; Zimerman, R.A.; Thompson, C.E. E484K as an innovative phylogenetic event for viral evolution: Genomic analysis of the E484K spike mutation in SARS-CoV-2 lineages from Brazil. Infect. Genet. Evol. 2021, 93, 104941. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- WHO. COVID-19 Weekly Epidemiological Update. Edition 91, Published 11 May 2022. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---11-may-2022 (accessed on 14 May 2022).
- Jaspe, R.C.; Loureiro, C.L.; Sulbaran, Y.; Moros, Z.C.; D’Angelo, P.; Rodríguez, L.; Zambrano, J.L.; Hidalgo, M.; Vizzi, E.; Alarcon, V.; et al. Introduction and rapid dissemination of SARS-CoV-2 Gamma Variant of Concern in Venezuela. Infect. Genet. Evol. 2021, 96, 105147. [Google Scholar] [CrossRef]
- Jaspe, R.C.; Zambrano, J.L.; Hidalgo, M.; Sulbaran, Y.; Loureiro, C.L.; Moros, Z.C.; Garzaro, D.J.; Liprandi, F.; Rangel, H.R.; Pujol, F.H. Detection of the Omicron variant of SARS-CoV-2 by restriction analysis targeting the mutations K417N and N440K of the Spike protein. Investig. Clin. 2022, 63, 92–99. [Google Scholar] [CrossRef]
- GISAID. SARS-CoV-2 (hCoV-19) Mutation Reports. Lineage|Mutation Tracker. Available online: https://outbreak.info/situation-reports?pango (accessed on 14 May 2022).
- Tatsi, E.B.; Filippatos, F.; Michos, A. SARS-CoV-2 variants and effectiveness of vaccines: A review of current evidence. Epidemiol. Infect. 2021, 149, e237. [Google Scholar] [CrossRef]
- Hu, Y.F.; Hu, J.C.; Gong, H.R.; Danchin, A.; Sun, R.; Chu, H.; Hung, I.F.; Yuen, K.Y.; To, K.K.; Zhang, B.Z.; et al. Computation of Antigenicity Predicts SARS-CoV-2 Vaccine Breakthrough Variants. Front. Immunol. 2022, 13, 861050. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Rojas, C.; Jimenez-Vasquez, V.; Hurtado, V.; Mestanza, O.; Molina, I.S.; Barcena, L.; Morales Ruiz, S.; Acedo, S.; Lizarraga, W.; Bailon, H.; et al. Genomic analysis reveals a rapid spread and predominance of lambda (C.37) SARS-COV-2 lineage in Peru despite circulation of variants of concern. J. Med. Virol. 2021, 93, 6845–6849. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Díaz, D.A.; Muñoz, A.L.; Tavera-Rodríguez, P.; Herrera-Sepúlveda, M.T.; Ruiz-Moreno, H.A.; Laiton-Donato, K.; Franco-Muñoz, C.; Pelaez-Carvajal, D.; Cuellar, D.; Muñoz-Suarez, A.M.; et al. Low Neutralizing Antibody Titers against the Mu Variant of SARS-CoV-2 in 31 BNT162b2 Vaccinated Individuals in Colombia. Vaccines 2022, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Kimura, I.; Shirakawa, K.; Takaori-Kondo, A.; Nakada, T.A.; Kaneda, A.; Nakagawa, S.; Sato, K. Genotype to Phenotype Japan (G2P-Japan) Consortium. Neutralization of the SARS-CoV-2 Mu Variant by Convalescent and Vaccine Serum. N. Engl. J. Med. 2021, 385, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ortiz, J.; Cardona, A.; Ciuoderis, K.; Averhoff, F.; Maya, M.A.; Cloherty, G.; Osorio, J.E. Assessment of SARS-CoV-2 Mu Variant Emergence and Spread in Colombia. JAMA Netw. Open 2022, 5, e224754. [Google Scholar] [CrossRef]
- Jaspe, R.C.; Sulbaran, Y.; Loureiro, C.L.; Moros, Z.C.; Marulanda, E.; Bracho, F.; Ramírez, N.A.; Canonico, Y.; D’Angelo, P.; Rodríguez, L.; et al. Detection of the Omicron variant of SARS-CoV-2 in international travelers returning to Venezuela. Travel Med. Infect. Dis. 2022, 48, 102326. [Google Scholar] [CrossRef]
- PAHO. COVID-19 Vaccination in the Americas. Available online: https://ais.paho.org/imm/IM_DosisAdmin-Vacunacion.asp (accessed on 14 May 2022).
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The Evolution and Biology of SARS-CoV-2 Variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef]
- Schwalb, A.; Armyra, E.; Méndez-Aranda, M.; Ugarte-Gil, C. COVID-19 in Latin America and the Caribbean: Two years of the pandemic. J. Intern. Med. 2022, in press. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Variant | Alpha | Beta | Gamma | Delta | Omicron * | Lambda | Mu |
|---|---|---|---|---|---|---|---|
| D405 | N (BA.2/3/4/5) | ||||||
| R408 | S (BA.2/5) | ||||||
| K417 | N | T | N (AY.1) ** | N | |||
| N440 | K | ||||||
| G446 | S (BA.1) | ||||||
| L452 | R | R (BA.4/5) | Q | ||||
| T478 | K | K | |||||
| E484 | K | K | A | K | |||
| F490 | S | ||||||
| N501 | Y | Y | Y | Y | Y |
| Variant | NGS | Sanger |
|---|---|---|
| Alpha | 11 | 11 |
| Gamma | 54 | 52 * |
| Delta | 163 (several sublineages) ** | 163 |
| Omicron | 61 *** | 61 |
| Lambda | 8 | 8 |
| Mu | 82 | 82 |
| Other lineages | 11 | 11 |
| Correlation | 388/390 (99.5%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaspe, R.C.; Loureiro, C.L.; Sulbaran, Y.; Moros, Z.C.; D’Angelo, P.; Hidalgo, M.; Rodríguez, L.; Alarcón, V.; Aguilar, M.; Sánchez, D.; et al. Description of a One-Year Succession of Variants of Interest and Concern of SARS-CoV-2 in Venezuela. Viruses 2022, 14, 1378. https://doi.org/10.3390/v14071378
Jaspe RC, Loureiro CL, Sulbaran Y, Moros ZC, D’Angelo P, Hidalgo M, Rodríguez L, Alarcón V, Aguilar M, Sánchez D, et al. Description of a One-Year Succession of Variants of Interest and Concern of SARS-CoV-2 in Venezuela. Viruses. 2022; 14(7):1378. https://doi.org/10.3390/v14071378
Chicago/Turabian StyleJaspe, Rossana C., Carmen L. Loureiro, Yoneira Sulbaran, Zoila C. Moros, Pierina D’Angelo, Mariana Hidalgo, Lieska Rodríguez, Víctor Alarcón, Marwan Aguilar, Doneyla Sánchez, and et al. 2022. "Description of a One-Year Succession of Variants of Interest and Concern of SARS-CoV-2 in Venezuela" Viruses 14, no. 7: 1378. https://doi.org/10.3390/v14071378
APA StyleJaspe, R. C., Loureiro, C. L., Sulbaran, Y., Moros, Z. C., D’Angelo, P., Hidalgo, M., Rodríguez, L., Alarcón, V., Aguilar, M., Sánchez, D., Ramírez, J., Garzaro, D. J., Zambrano, J. L., Liprandi, F., Rangel, H. R., & Pujol, F. H. (2022). Description of a One-Year Succession of Variants of Interest and Concern of SARS-CoV-2 in Venezuela. Viruses, 14(7), 1378. https://doi.org/10.3390/v14071378