
Sequencing during Times of Change: Evaluating SARS-CoV-2 Clinical Samples during the Transition from the Delta to Omicron Wave

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Extraction
2.2. Evaluating SARS-CoV-2 Viral Load in Patient Samples Using RT-qPCR
2.3. Sequencing Library Generation and Sequencing
2.4. Bioinformatics Analysis Flow
2.5. Read Frequency Cut off
2.6. Evaluating a Sample Specific ORF7a Region Deletion Using RT-PCR and Sanger Sequencing
2.7. Mutation and Statistical Analysis
3. Results
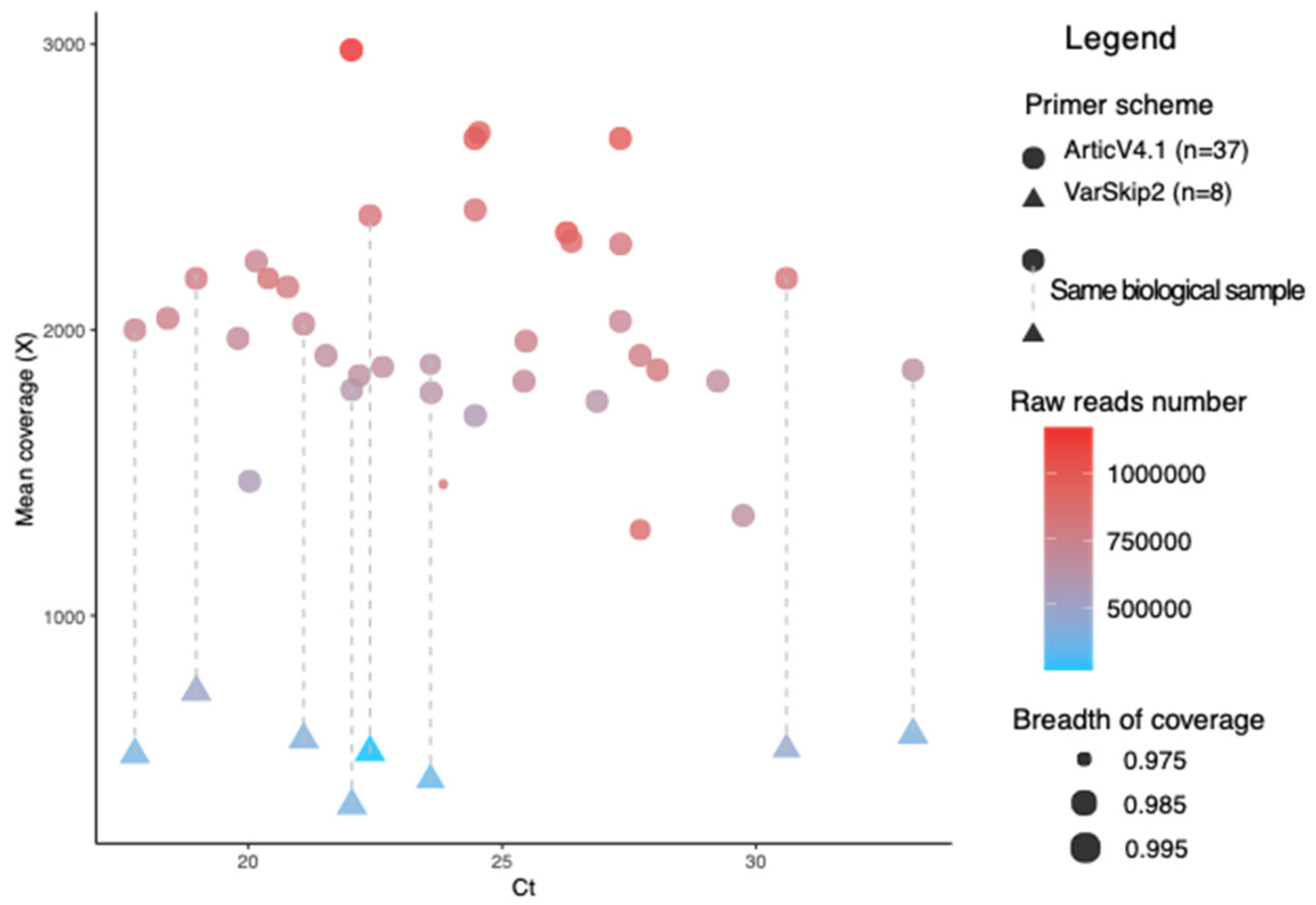
3.1. Evaluating SARS-CoV-2 Genomic RNA in Nasal-Pharyngeal Swabs from Patients with Breakthrough Infections
3.2. Bioinformatics Analysis of the SARS-CoV-2 Genomes Identifies Lineages and Sublineages
3.3. Assessment of Sequencing Outcomes Reveals a Sample-Specific Deletion in a Delta Variant
3.4. Analysis of Unique Mutations in Omicron and Delta Samples on Consensus Sequence Level
3.5. Analysis of Delta and Omicron Sequences for Evidence of Recombination Events and Association of Mutations with Disease Severity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 29 April 2022).
- World Health Organization (WHO). Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health; World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240018440.
- World Health Organization (WHO). Guidance for Surveillance of SARS-CoV-2 Variants: Interim Guidance, 9 August 2021; World Health Organization: Geneva, Switzerland, 2021.
- Kamar, N.; Abravanel, F.; Marion, O.; Couat, C.; Izopet, J.; Del Bello, A. Three Doses of an MRNA COVID-19 Vaccine in Solid-Organ Transplant Recipients. N. Engl. J. Med. 2021, 385, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Boyarsky, B.J.; Werbel, W.A.; Avery, R.K.; Tobian, A.A.R.; Massie, A.B.; Segev, D.L.; Garonzik-Wang, J.M. Antibody Response to 2-Dose SARS-CoV-2 MRNA Vaccine Series in Solid Organ Transplant Recipients. JAMA 2021, 325, 2204–2206. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Tenforde, M.W.; Gaglani, M.; Talbot, H.K.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Casey, J.D.; Mohr, N.M.; et al. MRNA Vaccine Effectiveness Against COVID-19 Hospitalization Among Solid Organ Transplant Recipients. J. Infect. Dis. 2022, jiac118. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell 2020, 183, 1901–1912.e9. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.; Regan, J.; Sparks, J.; Padera, R.; Qiu, X.; Solomon, I.; Kuo, H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 Evolution during Treatment of Chronic Infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- CDC Science Brief: Omicron (B.1.1.529) Variant. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html (accessed on 4 May 2022).
- Christoph, J.; Dorota, K.; Lennart, K.; Fabian, Z.; Timo, J.; Sparrer, K.M.J.; Frank, K. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e02077-21. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2022, 12. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H.; et al. Attenuated Replication and Pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- McMahan, K.; Giffin, V.; Tostanoski, L.H.; Chung, B.; Siamatu, M.; Suthar, M.S.; Halfmann, P.; Kawaoka, Y.; Piedra-Mora, C.; Jain, N.; et al. Reduced Pathogenicity of the SARS-CoV-2 Omicron Variant in Hamsters. Med 2022, 3, 262–268.e4. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron Virus Causes Attenuated Disease in Mice and Hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Fall, A.; Eldesouki, R.E.; Sachithanandham, J.; Morris, C.P.; Norton, J.M.; Gaston, D.C.; Forman, M.; Abdullah, O.; Gallagher, N.; Li, M.; et al. The Displacement of the SARS-CoV-2 Variant Delta with Omicron: An Investigation of Hospital Admissions and Upper Respiratory Viral Loads. eBioMedicine 2022, 79, 104008. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Wohlfahrt, J.; Bhatt, S.; Stegger, M.; Legarth, R.; Møller, C.H.; Skov, R.L.; Valentiner-Branth, P.; Voldstedlund, M.; Fischer, T.K.; et al. Risk of Hospitalisation Associated with Infection with SARS-CoV-2 Omicron Variant versus Delta Variant in Denmark: An Observational Cohort Study. Lancet Infect. Dis. 2022, 22, 967–976. [Google Scholar] [CrossRef]
- Sheikh, A.; Kerr, S.; Woolhouse, M.; McMenamin, J.; Robertson, C.; Simpson, C.R.; Millington, T.; Shi, T.; Agrawal, U.; Shahul Hameed, S.; et al. Severity of Omicron Variant of Concern and Effectiveness of Vaccine Boosters against Symptomatic Disease in Scotland (EAVE II): A National Cohort Study with Nested Test-Negative Design. Lancet Infect. Dis. 2022, 22, 959–966. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-Host Diversity and Transmission. Science 2021, 372, eabg0821. [Google Scholar] [CrossRef]
- Chan, A.E.R.; Jones, L.D.; Linger, M.; Kovach, J.D.; Torres-teran, M.M.; Wertz, A.; Donskey, C.J.; Peter, A. COVID-19 Infection and Transmission Includes Complex Sequence Diversity Abbreviated Title: SARS-CoV-2 Infection and Transmission Complexity. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.; Lensing, S.V.; Quail, M.A.; et al. Patterns of Within-Host Genetic Diversity in SARS-CoV-2. Elife 2021, 10, e66857. [Google Scholar] [CrossRef] [PubMed]
- Valesano, A.L.; Rumfelt, K.E.; Dimcheff, D.E.; Blair, C.N.; Fitzsimmons, W.J.; Petrie, J.G.; Martin, E.T.; Lauring, A.S. Temporal Dynamics of SARS-CoV-2 Mutation Accumulation within and across Infected Hosts. PLoS Pathog. 2021, 17, e1009499. [Google Scholar] [CrossRef]
- Braun, K.M.; Moreno, G.K.; Wagner, C.; Accola, M.A.; Rehrauer, W.M.; Baker, D.A.; Koelle, K.; O’Connor, D.H.; Bedford, T.; Friedrich, T.C.; et al. Acute SARS-CoV-2 Infections Harbor Limited within-Host Diversity and Transmit via Tight Transmission Bottlenecks. PLoS Pathog. 2021, 17, e1009849. [Google Scholar] [CrossRef]
- CDC. Real-Time RT-PCR Diagnostic Panel for Emergency Use Only; CDC: Atlanta, GA, USA, 2020; Volume 3.
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Castellano, S.; Cestari, F.; Faglioni, G.; Tenedini, E.; Marino, M.; Artuso, L.; Manfredini, R.; Luppi, M.; Trenti, T.; Tagliafico, E. iVar, an Interpretation-oriented Tool to Manage the Update and Revision of Variant Annotation and Classification. Genes 2021, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021; Volume 2, ISBN 3900051070. [Google Scholar]
- De Cáceres, M.; Legendre, P. Associations between Species and Groups of Sites: Indices and Statistical Inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef]
- Deng, X.; Evdokimova, M.; O’Brien, A.; Rowe, C.L.; Clark, N.M.; Harrington, A.; Reid, G.E.; Uprichard, S.L.; Baker, S.C. Breakthrough Infections with Multiple Lineages of SARS-CoV-2 Variants Reveals Continued Risk of Severe Disease in Immunosuppressed Patients. Viruses 2021, 13, 1743. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.-W. Emerging Vaccine-Breakthrough SARS-CoV-2 Variants. ACS Infect. Dis. 2022, 8, 546–556. [Google Scholar] [CrossRef]
- McEwen, A.E.; Cohen, S.; Bryson-Cahn, C.; Liu, C.; Pergam, S.A.; Lynch, J.; Schippers, A.; Strand, K.; Whimbey, E.; Mani, N.S.; et al. Variants of Concern Are Overrepresented Among Postvaccination Breakthrough Infections of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Washington State. Clin. Infect. Dis. 2022, 74, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Draper, J.; Gall, M.; Sim, E.M.; Arnott, A.; Agius, J.E.; Johnson-Mackinnon, J.; Fong, W.; Martinez, E.; Drew, A.P.; et al. Co-Infection with SARS-CoV-2 Omicron and Delta Variants Revealed by Genomic Surveillance. Nat. Commun. 2022, 13, 2745. [Google Scholar] [CrossRef] [PubMed]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Gray, K.; Gall, M.; Sadsad, R.; Arnott, A.; Johnson-Mackinnon, J.; Fong, W.; Basile, K.; Kok, J.; Dwyer, D.; et al. SARS-CoV-2 Genome Sequencing Methods Differ in Their Abilities To Detect Variants from Low-Viral-Load Samples. J. Clin. Microbiol. 2022, 59, e01046-21. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.; Huang, M.L.; Jerome, K.R.; Greninger, A.L. Identification of Multiple Large Deletions in ORF7a Resulting in In-Frame Gene Fusions in Clinical SARS-CoV-2 Isolates. J. Clin. Virol. 2020, 129, 104523. [Google Scholar] [CrossRef]
- Tse, H.; Wong, S.C.-Y.; Ip, K.-F.; Cheng, V.C.-C.; To, K.K.-W.; Lung, D.C.; Choi, G.K.-Y. Genome Sequences of Three SARS-CoV-2 ORF7a Deletion Variants Obtained from Patients in Hong Kong. Microbiol. Resour. Announc. 2021, 10, e00251-21. [Google Scholar] [CrossRef]
- Masters, P.S.; Rottier, P.J.M. Coronavirus Reverse Genetics by Targeted RNA Recombination. Curr. Top. Microbiol. Immunol. 2005, 287, 133–159. [Google Scholar] [CrossRef] [Green Version]
- Bolze, A.; White, S.; Basler, T.; Rossi, A.D.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Wyman, D.; Kil, E.; Dai, H.; et al. Evidence for SARS-CoV-2 Delta and Omicron Co-Infections and Recombination. medRxiv 2022. [Google Scholar] [CrossRef]
- Abbas, Q.; Kusakin, A.; Sharrouf, K.; Jyakhwo, S.; Komissarov, A.S. Follow-up Investigation and Detailed Mutational Characterization of the SARS-CoV-2 Omicron Variant Lineages (BA.1, BA.2, BA.3 and BA.1.1). bioRxiv 2022. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the Origin and Continuing Evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Du, P.; Yang, L.; Zhang, J.; Song, C.; Chen, D.; Song, Y.; Ding, N.; Hua, M.; Han, K.; et al. Two-Step Fitness Selection for Intra-Host Variations in SARS-CoV-2. Cell Rep. 2022, 38, 110205. [Google Scholar] [CrossRef]
- CDC. Science Brief: Evidence Used to Update the List of Underlying Medical Conditions Associated with Higher Risk for Severe COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/underlying-evidence-table.html (accessed on 10 May 2022).
- Davies, N.G.; Klepac, P.; Liu, Y.; Prem, K.; Jit, M.; Pearson, C.A.B.; Quilty, B.J.; Kucharski, A.J.; Gibbs, H.; Clifford, S.; et al. Age-Dependent Effects in the Transmission and Control of COVID-19 Epidemics. Nat. Med. 2020, 26, 1205–1211. [Google Scholar] [CrossRef]
- Drucker, D.J. Diabetes, Obesity, Metabolism, and SARS-CoV-2 Infection: The End of the Beginning. Cell Metab. 2021, 33, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Simons, L.M.; Lorenzo-Redondo, R.; Gibson, M.; Kinch, S.L.; Vandervaart, J.P.; Reiser, N.L.; Eren, M.; Lux, E.; McNally, E.M.; Tambur, A.R.; et al. Assessment of Virological Contributions to COVID-19 Outcomes in a Longitudinal Cohort of Hospitalized Adults. Open Forum Infect. Dis. 2022, 9, ofac027. [Google Scholar] [CrossRef] [PubMed]
- CDC. Risk for COVID-19 Infection, Hospitalization, and Death By Age Group. Available online: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/hospitalization-death-by-age.html (accessed on 10 May 2022).
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. MBio 2022, 13, e02979-21. [Google Scholar] [CrossRef] [PubMed]
- CDC Science Brief: SARS-CoV-2 Infection-Induced and Vaccine-Induced Immunity. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/vaccine-induced-immunity.html (accessed on 29 April 2022).
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in Older Adults: What We May Expect Regarding Pathogenesis, Immune Responses, and Outcomes. GeroScience 2020, 42, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, H.; Teran, R.A.; Barbian, H.J.; Love, S.; Berg, R.; Black, S.R.; Ghinai, I.; Kerins, J.L.; Patrick, S.; Kauerauf, J.; et al. Multistate Outbreak of Infection with SARS-CoV-2 Omicron Variant after Event in Chicago, Illinois, USA, 2021. Emerg Infect Dis. 2022, 28, 1281–1283. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e00261-21. [Google Scholar] [CrossRef]
- Al Khatib, H.A.; Benslimane, F.M.; Elbashir, I.E.; Coyle, P.V.; Al Maslamani, M.A.; Al-Khal, A.; Al Thani, A.A.; Yassine, H.M. Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients With Variable Disease Severities. Front. Cell. Infect. Microbiol. 2020, 10, 575613. [Google Scholar] [CrossRef]
- Abdullah, M.S.; Taha, Y.T.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid Assessment of SARS-CoV-2–Evolved Variants Using Virus-like Particles. Science. 2021, 374, 1626–1632. [Google Scholar] [CrossRef]
- Holland, L.A.; Kaelin, E.A.; Maqsood, R.; Estifanos, B.; Wu, L.I.; Varsani, A.; Halden, R.U.; Hogue, B.G.; Scotch, M.; Lim, E.S. An 81-Nucleotide Deletion in SARS-CoV-2 ORF7a Identified from Sentinel Surveillance in Arizona (January to March 2020). J. Virol. 2020, 94, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Nichols, J.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; Lee, H.; Vanderwood, K.K.; Bimczok, D.; et al. SARS-CoV-2 Genomic Surveillance Identifies Naturally Occurring Truncation of ORF7a That Limits Immune Suppression. Cell Rep. 2021, 35, 109197. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a Promotes Antagonism of Interferon Response. Cell. Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.L.; Fleming, J.O.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Baker, S.C. Generation of Coronavirus Spike Deletion Variants by High-Frequency Recombination at Regions of Predicted RNA Secondary Structure. J. Virol. 1997, 71, 6183–6190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, R.C.A.; Mahadeshwar, G.; Wan, H.; Huston, N.C.; Pyle, A.M. The Global and Local Distribution of RNA Structure throughout the SARS-CoV-2 Genome. J. Virol. 2022, 95, e02190-20. [Google Scholar] [CrossRef] [PubMed]
- Proposal to Split B.1.1.529 to Incorporate a Newly Characterised Sibling Lineage. Available online: https://github.com/cov-lineages/pango-designation/issues/361 (accessed on 20 April 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Primer Sequence (5′ to 3′) |
|---|---|
| PCR pF | AACACAGACCATTCCAGTAGC |
| PCR pR | GACACGGGTCATCAACTACAT |
| Sequencing Primer pF1 | CACTGATAACACTCGCTACTTG |
| Sequencing Primer pF2 | GCTTTGCTTGTACAGTAAGTGAC |
| Sequencing Primer pR3 | TGCAGCTACAGTTGTGATGAT |
| Sequencing Primer pR4 | TGCAGTTCAAGTGAGAACCA |
| N or Range | % or Median | ||
|---|---|---|---|
| Sex | Male | 20 | 59% |
| Female | 14 | 41% | |
| Age Range (Median) | Male | 30–79 | 55 |
| Female | 30–67 | 62.5 | |
| Vaccination Status | Vaccinated (≥1 dose) | 33 | 97% |
| Unvaccinated | 1 | 3% | |
| Immunosuppression Status | Immunosuppressed * | 27 | 79% |
| Normal | 7 | 21% | |
| Hospitalization Status ** | Hospitalized | 28 | 82% |
| Not hospitalized | 6 | 18% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, S.; Ali, M.S.; Evdokimova, M.; Reid, G.E.; Clark, N.M.; Uprichard, S.L.; Baker, S.C. Sequencing during Times of Change: Evaluating SARS-CoV-2 Clinical Samples during the Transition from the Delta to Omicron Wave. Viruses 2022, 14, 1408. https://doi.org/10.3390/v14071408
Feng S, Ali MS, Evdokimova M, Reid GE, Clark NM, Uprichard SL, Baker SC. Sequencing during Times of Change: Evaluating SARS-CoV-2 Clinical Samples during the Transition from the Delta to Omicron Wave. Viruses. 2022; 14(7):1408. https://doi.org/10.3390/v14071408
Chicago/Turabian StyleFeng, Shuchen, Mudassir S. Ali, Monika Evdokimova, Gail E. Reid, Nina M. Clark, Susan L. Uprichard, and Susan C. Baker. 2022. "Sequencing during Times of Change: Evaluating SARS-CoV-2 Clinical Samples during the Transition from the Delta to Omicron Wave" Viruses 14, no. 7: 1408. https://doi.org/10.3390/v14071408
APA StyleFeng, S., Ali, M. S., Evdokimova, M., Reid, G. E., Clark, N. M., Uprichard, S. L., & Baker, S. C. (2022). Sequencing during Times of Change: Evaluating SARS-CoV-2 Clinical Samples during the Transition from the Delta to Omicron Wave. Viruses, 14(7), 1408. https://doi.org/10.3390/v14071408