
Evolutionary Dynamics of Foot and Mouth Disease Virus Serotype A and Its Endemic Sub-Lineage A/ASIA/Iran-05/SIS-13 in Pakistan

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation
2.2. Genome Sequencing
2.3. Data Retrieval and Sequence Analysis
2.4. Recombination Analysis
2.5. Natural Selection Analysis
2.6. Phylogeographic Analysis
3. Results
3.1. Sequence Analysis
3.2. Recombination Analysis
3.3. Natural Selection Analysis
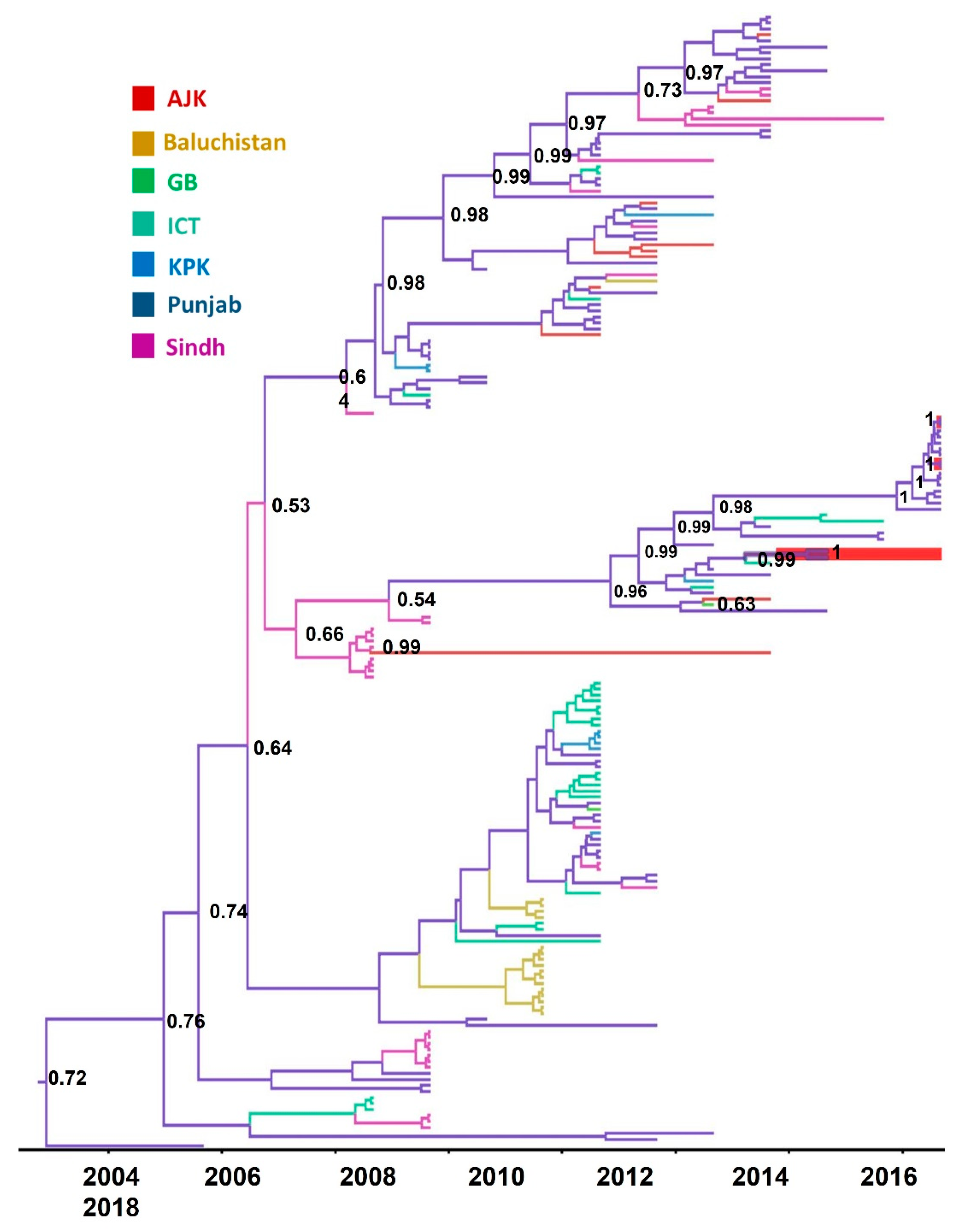
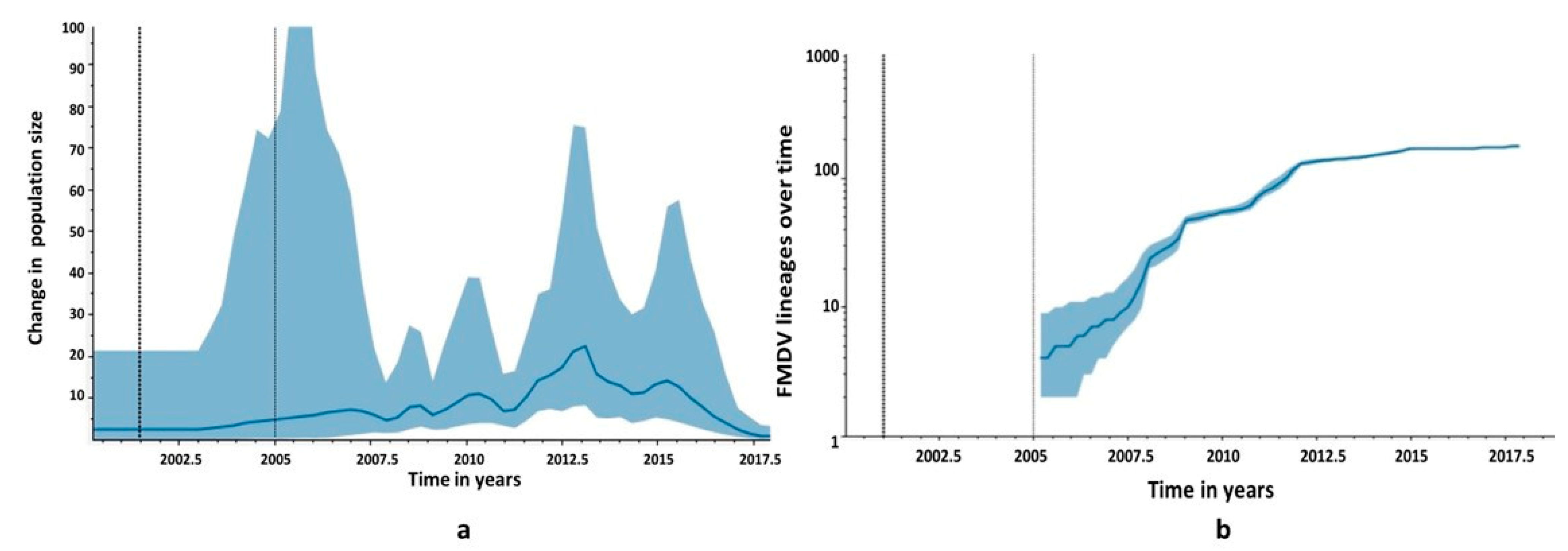
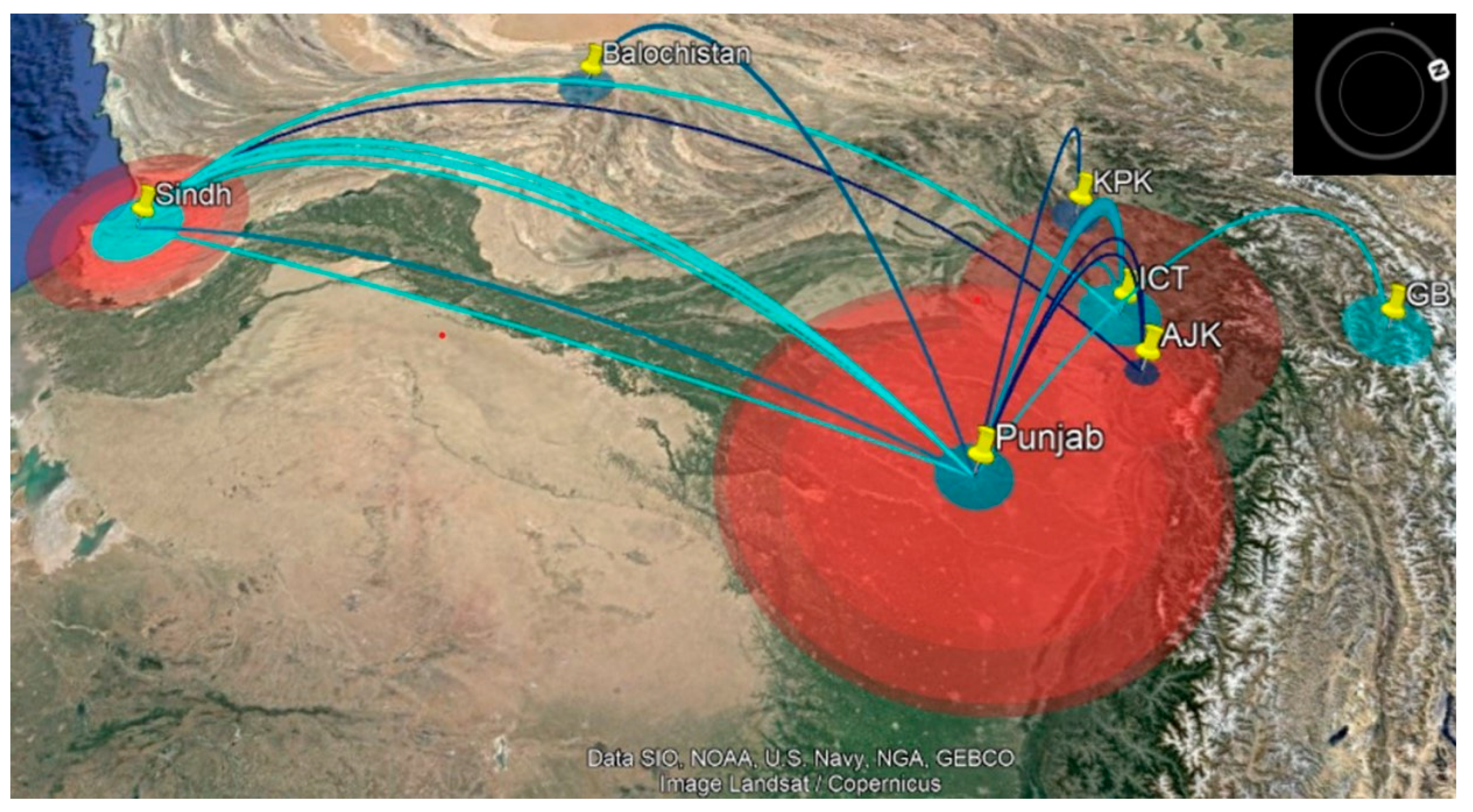
3.4. Phylogeographic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knowles, N.J.; Samuel, A.R.; Davies, P.R.; Midgley, R.J. Pandemic Strain of Foot-and-Mouth Disease Virus Serotype O. Emerg. Infect. Dis. 2005, 11, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Brito, B.P.; Rodriguez, L.L.; Hammond, J.M.; Pinto, J.; Perez, A.M. Review of the Global Distribution of Foot-and-Mouth Disease Virus from 2007 to 2014. Transbound. Emerg. Dis. 2015, 64, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Al-Hosary, A.A.; Kandeil, A.; El-Taweel, A.N.; Nordengrahn, A.; Merza, M.; Badra, R.; Kayali, G.; Ali, M.A. Co-infection with different serotypes of FMDV in vaccinated cattle in Southern Egypt. Virus Genes 2019, 55, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Pauszek, S.J.; Ludi, A.; LaRocco, M.; Khan, E.U.H.; Afzal, M.; Arshed, M.J.; Farooq, U.; Arzt, J.; Bertram, M.; et al. Genetic diversity and comparison of diagnostic tests for characterization of foot-and-mouth disease virus strains from Pakistan 2008–2012. Transbound. Emerg. Dis. 2018, 65, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Ferrari, G.; Ahmed, S.; Normann, P.; Belsham, G.J. Molecular characterization of serotype Asia-1 foot-and-mouth disease viruses in Pakistan and Afghanistan; emergence of a new genetic Group and evidence for a novel recombinant virus. Infect. Genet. Evol. 2011, 11, 2049–2062. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Ferrari, G.; Ahmed, S.; Normann, P.; Belsham, G.J. Genetic diversity of foot-and-mouth disease virus serotype O in Pakistan and Afghanistan, 1997–2009. Infect. Genet. Evol. 2011, 11, 1229–1238. [Google Scholar] [CrossRef]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative Genomics of Foot-and-Mouth Disease Virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, F.-P.; Knowles, N.J.; Wadsworth, J.; Knowles, N.J.; Wadsworth, J.; King, D.P. VP1 sequencing protocol for foot and mouth disease virus molecular epidemiology. Rev. Sci. Tech. 2017, 35, 741–755. [Google Scholar] [CrossRef] [Green Version]
- Grazioli, S.; Fallacara, F.; Brocchi, E. Mapping of antigenic sites of foot-and-mouth disease virus serotype Asia 1 and relationships with sites described in other serotypes. J. Gen. Virol. 2013, 94, 559–569. [Google Scholar] [CrossRef]
- Mahapatra, M.; Seki, C.; Upadhyaya, S.; Barnett, P.V.; La Torre, J.; Paton, D.J. Characterisation and epitope mapping of neutralising monoclonal antibodies to A24 Cruzeiro strain of FMDV. Vet. Microbiol. 2011, 149, 242–247. [Google Scholar] [CrossRef]
- Theys, K.; Libin, P.; Vandamme, A.; Nowe, A.; Abecasis, A.B. ScienceDirect The impact of HIV-1 within-host evolution on transmission dynamics. Curr. Opin. Virol. 2018, 1, 92–101. [Google Scholar] [CrossRef]
- Kustin, T.; Stern, A. Biased Mutation and Selection in RNA Viruses. Mol. Biol. Evol. 2021, 38, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.S.; Donaldson, A.I. Natural adaption to pigs of a Taiwanese isolate of foot-and-mouth disease virus. Vet. Rec. 1997, 141, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Maree, F.; de Klerk-Lorist, L.-M.; Gubbins, S.; Zhang, F.; Seago, J.; Pérez-Martín, E.; Reid, L.; Scott, K.; van Schalkwyk, L.; Bengis, R.; et al. Differential Persistence of Foot-and-Mouth Disease Virus in African Buffalo Is Related to Virus Virulence. J. Virol. 2016, 90, 5132–5140. [Google Scholar] [CrossRef] [Green Version]
- Brito, B.P.; Mohapatra, J.K.; Subramaniam, S.; Pattnaik, B.; Rodriguez, L.L.; Moore, B.R.; Perez, A.M.; Ibrahim, B.; Mcmahon, D.P.; Hufsky, F.; et al. Dynamics of widespread foot-and-mouth disease virus serotypes A, O and Asia-1 in southern Asia: A Bayesian phylogenetic perspective. Transbound. Emerg. Dis. 2018, 65, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.T.; Samuel, A.R.; Knowles, N.J. The generation and persistence of genetic variation in foot-and-mouth disease virus. Prev. Vet. Med. 2001, 51, 111–124. [Google Scholar] [CrossRef]
- El Damaty, H.M.; Fawzi, E.M.; Neamat-Allah, A.N.F.; Elsohaby, I.; Abdallah, A.; Farag, G.K.; El-Shazly, Y.A.; Mahmmod, Y.S. Characterization of foot and mouth disease virus serotype sat-2 in swamp water buffaloes (Bubalus bubalis) under the egyptian smallholder production system. Animals 2021, 11, 1697. [Google Scholar] [CrossRef] [PubMed]
- Government of Pakistan Finance Division. Pakistan Economic Survey 2017-18. Pakistan Econ. Surv. 2019; Chapter 2. Available online: https://ww.finance.gov.pk/survey/chapters_18/Economic_Survey_2017_18.pdf (accessed on 15 March 2022).
- Jamal, S.M.; Belsham, G.J. Infection, Genetics and Evolution Molecular epidemiology, evolution and phylogeny of foot-and-mouth disease virus P1-2A. Infect. Genet. Evol. 2018, 59, 84–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, H.; Khan, M.A.; Afzal, M.; Farooq, W.; Rani, S. Baseline Survey of Progressive Control of Foot & Mouth Disease Project in Pakistan. Front. Vet. Sci. 2014, 8, 703473. [Google Scholar]
- Di Nardo, A.; Knowles, N.J.; Paton, D.J. Combining livestock trade patterns with phylogenetics to help understand the spread of foot and mouth disease in sub-Saharan Africa, the Middle East and Southeast Asia. OIE Rev. Sci. Tech. 2011, 30, 63–85. [Google Scholar] [CrossRef]
- Di Nardo, A.; Ferretti, L.; Wadsworth, J.; Mioulet, V.; Gelman, B.; Karniely, S.; Scherbakov, A.; Ziay, G.; Özyörük, F.; Parlak, Ü.; et al. Evolutionary and Ecological Drivers Shape the Emergence and Extinction of Foot-and-Mouth Disease Virus Lineages. Mol. Biol. Evol. 2021, 38, 4346–4361. [Google Scholar] [CrossRef]
- Klein, J.; Hussain, M.; Ahmad, M.; Normann, P.; Afzal, M.; Alexandersen, S. Genetic characterisation of the recent foot-and-mouth disease virus subtype A/IRN/2005. Virol. J. 2007, 4, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Country Reports: 2020|World Reference Laboratory for Foot-and-Mouth Disease. Available online: http://www.wrlfmd.org/country-reports/country-reports-2020 (accessed on 15 March 2022).
- LaRocco, M.; Krug, P.W.; Kramer, E.; Ahmed, Z.; Pacheco, J.M.; Duque, H.; Baxt, B.; Rodriguez, L.L. A continuous bovine kidney cell line constitutively expressing bovine αVβ6 integrin has increased susceptibility to foot-and-mouth disease virus. J. Clin. Microbiol. 2013, 51, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, T.; Yamada, M.; Fukai, K.; Shimada, N.; Morioka, K.; Yoshida, K.; Sakamoto, K.; Kanno, T.; Yamakawa, M. Genome variability of foot-and-mouth disease virus during the short period of the 2010 epidemic in Japan. Vet. Microbiol. 2017, 199, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Maung, W.Y.; Nishi, T.; Kato, T.; Lwin, K.O.; Fukai, K. Genome Sequences of Foot-and-Mouth Disease Viruses of Serotype O Lineages Mya-98 and Ind-2001d Isolated from Cattle and Buffalo in Myanmar. Microbiol. Resour. Announc. 2019, 8, e01737-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Figtree v 1.4.4. Available online: http://tree.bio.ed.ac.uk/ (accessed on 27 November 2020).
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Datamonkey Adaptive Evolution Server. Available online: https://www.datamonkey.org/ (accessed on 3 January 2022).
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Grossman, Z.; Gravenor, M.B.; Richman, D.D.; Leigh Brown, A.J. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2006, 2, 0530–0538. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Knowles, N.J.; Nazem Shirazi, M.H.; Wadsworth, J.; Swabey, K.G.; Stirling, J.M.; Statham, R.J.; Li, Y.; Hutchings, G.H.; Ferris, N.P.; Parlak, Ü.; et al. Recent Spread of a New Strain (A-Iran-05) of Foot-and-Mouth Disease Virus Type A in the Middle East. Transbound. Emerg. Dis. 2009, 56, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Nazem Shirazi, M.H.; Ozyoruk, F.; Parlak, U.; Normann, P.; Belsham, G.J. Evidence for multiple recombination events within foot-and-mouth disease viruses circulating in West Eurasia. Transbound. Emerg. Dis. 2020, 67, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Jamal, S.M.; Romey, A.; Gorna, K.; Kakar, M.A.; Abbas, F.; Ahmad, J.; Zientara, S.; Bakkali Kassimi, L. Genetic Characterization of Serotypes A and Asia-1 Foot-and-mouth Disease Viruses in Balochistan, Pakistan, in 2011. Transbound. Emerg. Dis. 2017, 64, 1569–1578. [Google Scholar] [CrossRef]
- Upadhyaya, S.; Mahapatra, M.; Mioulet, V.; Parida, S. Molecular Basis of Antigenic Drift in Serotype O Foot-and-Mouth Disease Viruses (2013–2018) from Southeast Asia. Viruses 2021, 13, 1886. [Google Scholar] [CrossRef]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Lineage | No. of Taxa | VP2 (AA = 218) | VP3 (AA = 221) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acid Site No. | 38 | 64 | 79 | 98 | 86 | 133 | 134 | 195 | 207 | 3 | 59 | 65 | 70 | 92 | 94 | 95 | 99 | 117 | 159 | 175 | 220 | |
| AFG-07 | 1 Taxon | T | K | E | F | E | S | P | S | F | V | D | V | E | S | I | A | T | S | S | V | A |
| HER-10 | 16 Taxa | Q | S | P | L | N | E | |||||||||||||||
| BAR-08 | 2 Taxa | V | Y | D | A | |||||||||||||||||
| SIS-13 | MZ493232 | D | S | S | S | A | T | |||||||||||||||
| SIS-13 | MZ493233, MZ493234 | M | A | P | ||||||||||||||||||
| FAR-11 | 1 Taxon | A | T | A | ||||||||||||||||||
| FAR-09 | 1 Taxon | L | ||||||||||||||||||||
| ESF-10 | 6 Taxa | I | D | L | T | |||||||||||||||||
| SIN-08 | 4 Taxa | D | T | |||||||||||||||||||
| Sub-Lineage | No. of Taxa | VP1 (AA = 211) | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acid Site No. | 17 | 24 | 28 | 43 | 45 | 46 | 65 | 83 | 95 | 96 | 99 | 109 | 134 | 135 | 140 | 141 | 142 | 145 | 149 | 154 | 155 | 160 | 168 | 171 | 196 | 198 | 204 | |
| AFG-07 | 1 Taxon | N | A | H | N | V | S | L | D | V | G | A | K | S | K | S | G | G | G | S | V | A | S | R | T | S | Q | Q |
| HER-10 | 16 Taxa | G | K | |||||||||||||||||||||||||
| BAR-08 | 2 Taxa | D | V | A | F | E | E | M | N | G | R | |||||||||||||||||
| SIS-13 | MZ493232 | G | N | G | S | P | I | R | ||||||||||||||||||||
| SIS-13 | MZ493233 MZ493234 | Q | E | E | R | |||||||||||||||||||||||
| FAR-11 | 1 Taxon | T | A | E | R | G | I | L | S | |||||||||||||||||||
| FAR-09 | 1 Taxon | V | E | R | G | S | D | L | T | R | ||||||||||||||||||
| ESF-10 | 6 Taxa | T | Q | S | E | E | R | S | D | A | ||||||||||||||||||
| SIN-08 | 4 Taxa | R | G | Q | ||||||||||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naqvi, S.S.; Bostan, N.; Fukai, K.; Ali, Q.; Morioka, K.; Nishi, T.; Abubakar, M.; Ahmed, Z.; Sattar, S.; Javed, S.; et al. Evolutionary Dynamics of Foot and Mouth Disease Virus Serotype A and Its Endemic Sub-Lineage A/ASIA/Iran-05/SIS-13 in Pakistan. Viruses 2022, 14, 1634. https://doi.org/10.3390/v14081634
Naqvi SS, Bostan N, Fukai K, Ali Q, Morioka K, Nishi T, Abubakar M, Ahmed Z, Sattar S, Javed S, et al. Evolutionary Dynamics of Foot and Mouth Disease Virus Serotype A and Its Endemic Sub-Lineage A/ASIA/Iran-05/SIS-13 in Pakistan. Viruses. 2022; 14(8):1634. https://doi.org/10.3390/v14081634
Chicago/Turabian StyleNaqvi, Syeda Sumera, Nazish Bostan, Katsuhiko Fukai, Qurban Ali, Kazuki Morioka, Tatsuya Nishi, Muhammad Abubakar, Zaheer Ahmed, Sadia Sattar, Sundus Javed, and et al. 2022. "Evolutionary Dynamics of Foot and Mouth Disease Virus Serotype A and Its Endemic Sub-Lineage A/ASIA/Iran-05/SIS-13 in Pakistan" Viruses 14, no. 8: 1634. https://doi.org/10.3390/v14081634
APA StyleNaqvi, S. S., Bostan, N., Fukai, K., Ali, Q., Morioka, K., Nishi, T., Abubakar, M., Ahmed, Z., Sattar, S., Javed, S., Tariq, A., & Sadiq, A. (2022). Evolutionary Dynamics of Foot and Mouth Disease Virus Serotype A and Its Endemic Sub-Lineage A/ASIA/Iran-05/SIS-13 in Pakistan. Viruses, 14(8), 1634. https://doi.org/10.3390/v14081634