
Finding Asymptomatic Spreaders in a COVID-19 Transmission Network by Graph Attention Networks

Abstract
:1. Introduction
2. Methods
2.1. Background
2.1.1. COVID-19 Transmission Network Representation
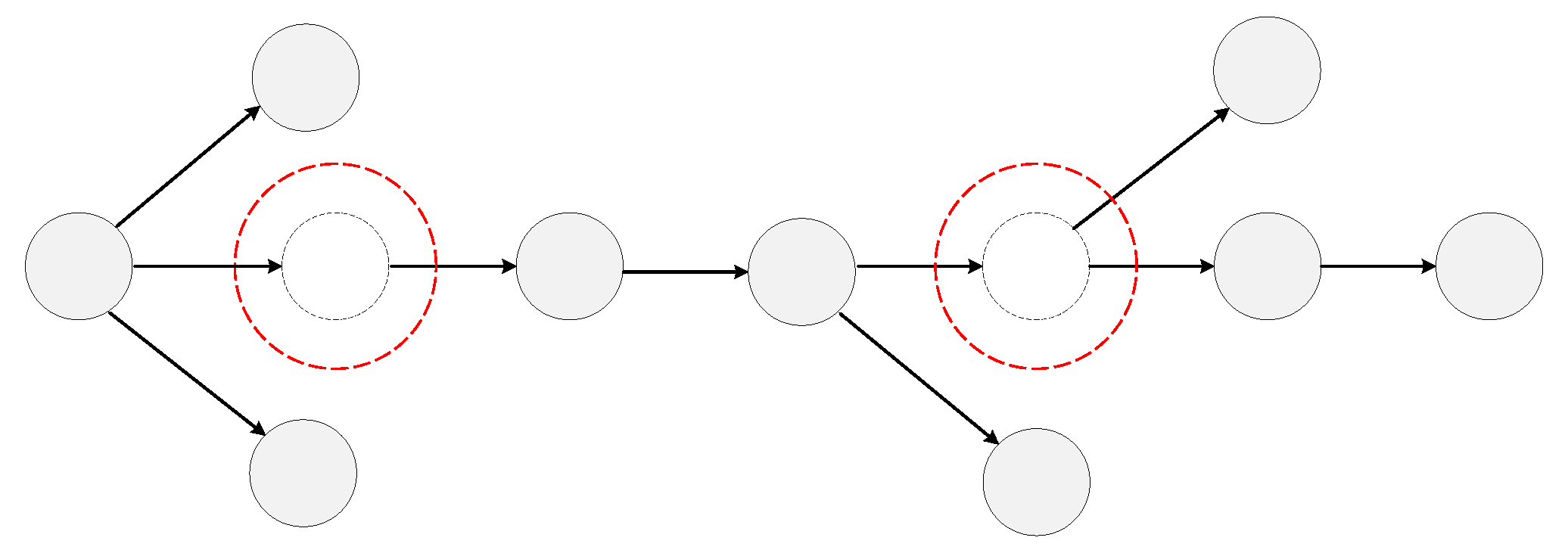
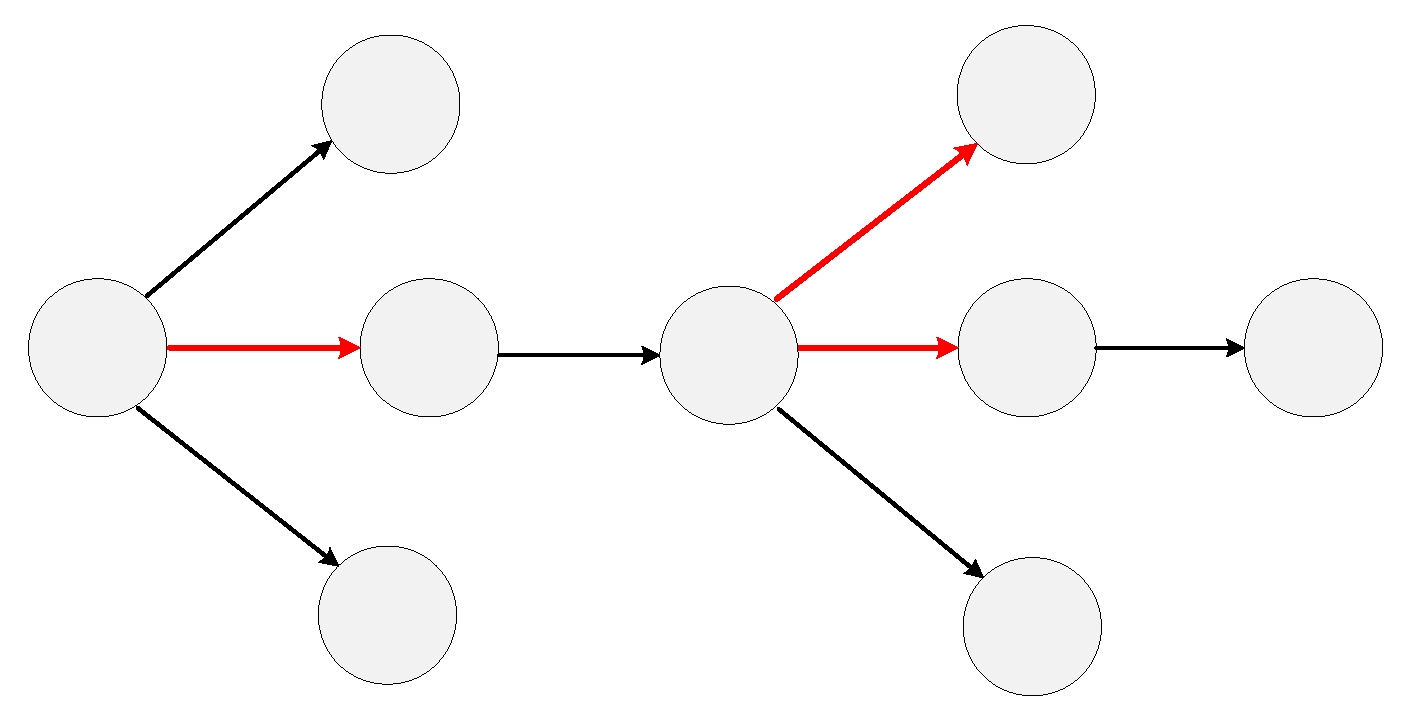
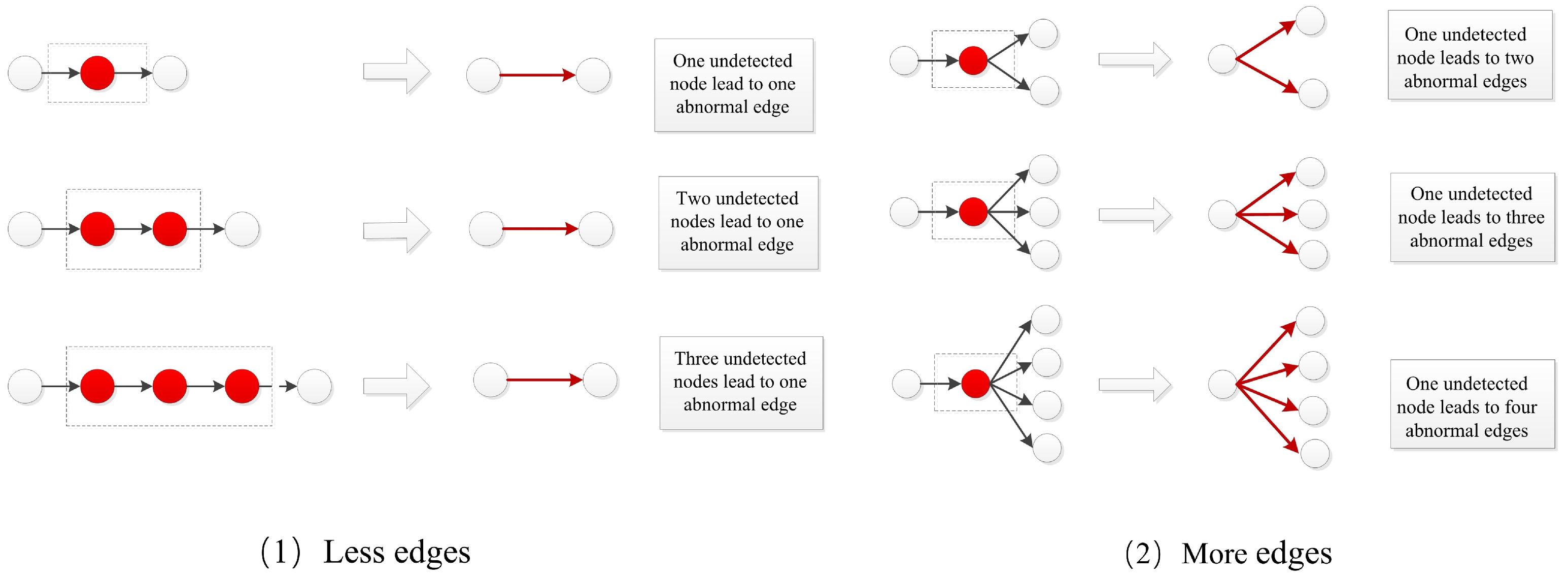
2.1.2. Undetected Nodes and Abnormal Edges
2.2. Model
2.2.1. Subgraph Extraction
2.2.2. Subgraph Representation
2.2.3. Information Aggregation Based on Graph Attention Mechanism
2.2.4. Graph Context Loss Function
2.2.5. Similarity Calculation
2.3. Experimental Design
2.3.1. Two Kinds of Test Experiments
2.3.2. Comparison Algorithms
2.3.3. Evaluation Metrics
3. Materials
3.1. Simulation Data
3.1.1. Training Data Generation
- Sequence simulation
- Transmission network simulation
| Algorithm 1 Generating procedure of simulated data. |
Input: Model parameters N, , L, p Output: Transmission network and the corresponding gene sequence whiledo randomly choose A, T, G or C end while Return gene sequence for node whiledo generate child nodes by choosing an R0 value from the range of R0 values following the probability for each value while do copy the gene sequence of node Randomly change the gene to generate a gene sequence for child nodes end while Return gene sequence of each node end while Return transmission network |
3.1.2. Test Data Generation
| Algorithm 2 Test transmission network and label data generation. |
Input: a transmission network, the proportion of removed nodes Output: a test network, label data set whiledo randomly choose nodes in the network end while if the selected node is at the margin of the network then retain it in the network else remove it from the network connect the parent node of the removed node to its child nodes Add new connected edges into the label set end if Return a test network and label data set |
3.2. Real Data
3.2.1. Data Resource
3.2.2. Real Data Processing
4. Results
4.1. Simulation Experiment Results
4.2. Real Experiment Results
5. Discussion
5.1. Training Details of Simulation Experiment
5.2. The Influence of Network Structure
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus disease 2019 |
| RVTR | RNA virus transmission network representation model |
| GCN | Graph convolutional neural network |
| SDNE | Structural deep network embedding |
| PCA | Principal component analysis |
| AE | Autoencoder |
Appendix A
Appendix A.1. Comparison Algorithm Description
- GCN uses a low pass filter to generate node embedding; the information of each node is transferred to the neighbor nodes in the graph, and the graph can transfer information layer by layer through a convolution operation.
- SDNE uses a deep neural network to model the nonlinearity between node representations. The whole model can be divided into two parts: the modeling module of level 1 similarity supervised by the Laplace matrix and the modeling of the level 2 similarity relation by an unsupervised deep autoencoder.
- PCA is one of the most widely used data dimension reduction algorithms. The main idea is to map the possibly correlated N-dimensional feature data to the linearly unrelated K-dimension through orthogonal change, reconstruct the K-dimensional features on the basis of the original N-dimensional features, and then achieve efficient and accurate representation of the original data. The transformed K-dimensional features are called principal components.
- AE consists of an encoder and a decoder. The encoder can compress the input attributes into potential spatial representations, and the decoder can reconstruct the data input from potential spatial representations. Autoencoders are often used for dimensionality reduction or feature learning.
- DIRECT is to use the gene sequence directly to calculate node similarity. After the gene sequence of each node was encoded by a one-hot encoder, it was directly used to calculate the similarity score for each pair node without dimension reduction.
Appendix A.2. Reproducibility
Appendix A.3. Real Data Description
- Data resource: Both FASTA sequence data and the corresponding epidemiological data of Canadian, New Zealand, New York State, and Australia were downloaded from GISAID (https://www.gisaid.org/ Accessed on 1 September 2020.) [26]. We confirmed that the selected data had complete sequences with high sequence coverage. The length of complete sequence comprised of genoms is greater than 29,000, the proportion of undefined bases for high coverage sequences was less than , while it was greater than for low coverage sequences.
- Data description: The data collection of the Canadian dataset is from January 2020 to July 2020, and there was a high sampling rate with over 4 million tests being performed (with a weekly average of over 55,000 tests at positivity), thus ensuring that the dataset is concise. The sampling time of the Australia dataset was from January 2020 to April 2020. The sampling date of New Zealand is from March 2020 to January 2021, and the sampling time span was the longest in these datasets. However, half of the sampled infections were detected in March and April of 2020, and there were quite few infections sampled in May, June and July of 2020. This spread is unique when compared with other transmission datasets. During the study period (February 2020 to September 2020), New York State was considered a COVID-19 epicenter with the second-highest case numbers of COVID-19 in the USA in March 2020; however, it had a lower sampling rate in comparison to its rate of disease incidence [10]. The sampling time of the Alberta dataset was from March 2020 to May 2020.
Appendix A.4. Real Data Process
- Sequence generation for patient “0”As the transmission network inferred by TransPhylo was based on the assumption that all infections have a common ancestor, we needed to simulate the gene sequence of patient “0” for each inferred transmission network before we trained and tested our model on them. If we removed patient “0” from the network, the transmission network collapsed into several disconnected small networks. The basic idea to simulate the sequence for patients “0” was also based on the mutation of SARS-CoV-2 virus; all of the individuals in a local module in a network share a common quasi-species signature of the gene sequence. We sought the nearest neighbor node of patient “0” in the inferred transmission network, then copied its sequence and changed some genes randomly to reflect the difference between patient “0” and its child nodes.
- Sequence alignment for different test dataThe length of the sequence in the FASTA data is slightly different from dataset to dataset, and the different sequence data need to be adjusted for alignment before being used in the experiments. As our trained model is based on the Australia dataset, we used ‘-’ to fill the sequence whose length is shorter than that of Aussies. For the sequences in a common dataset, the number and location of newly added genes ‘-’ were the same, so this operation had no effect on the test dataset.
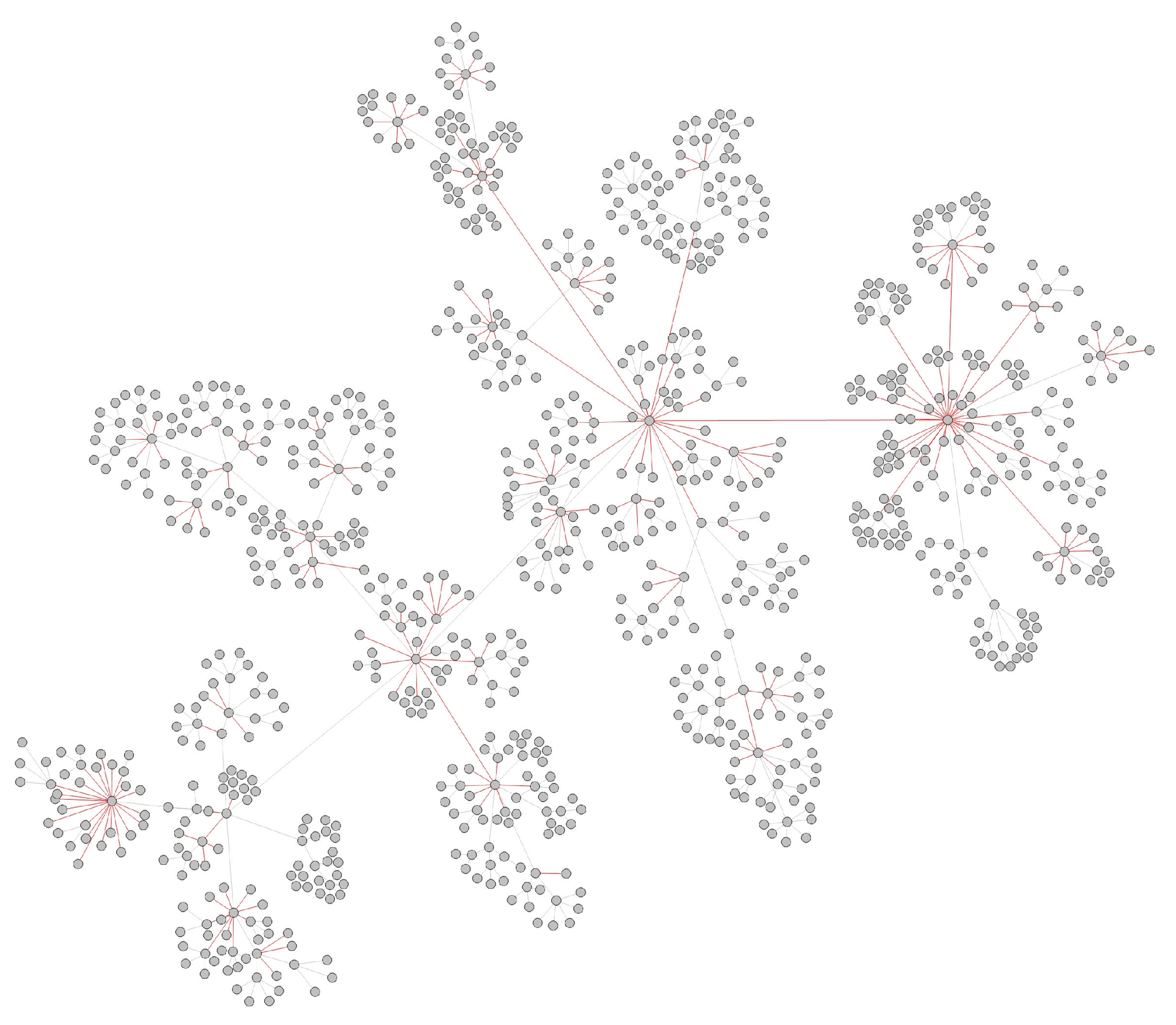
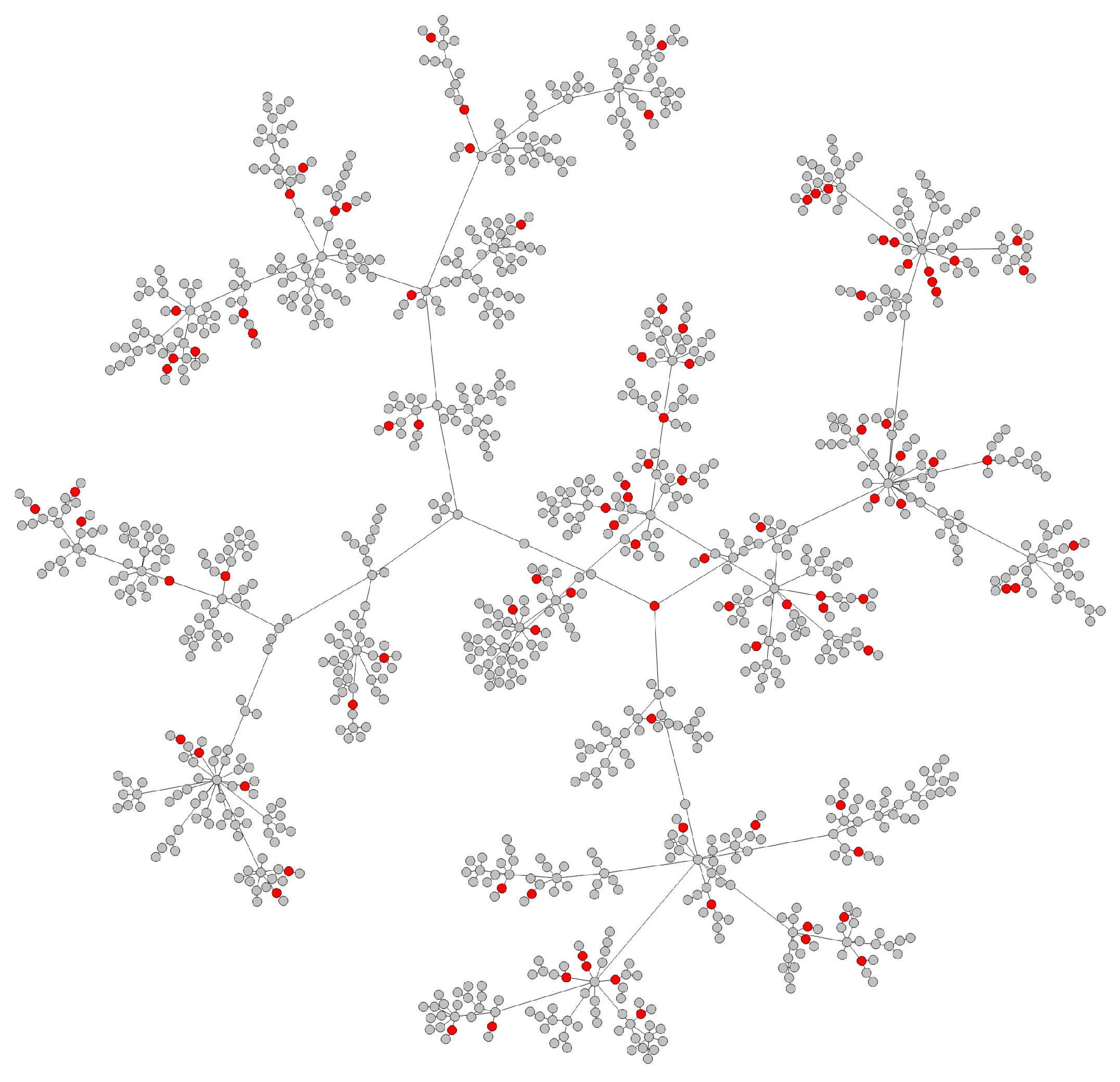
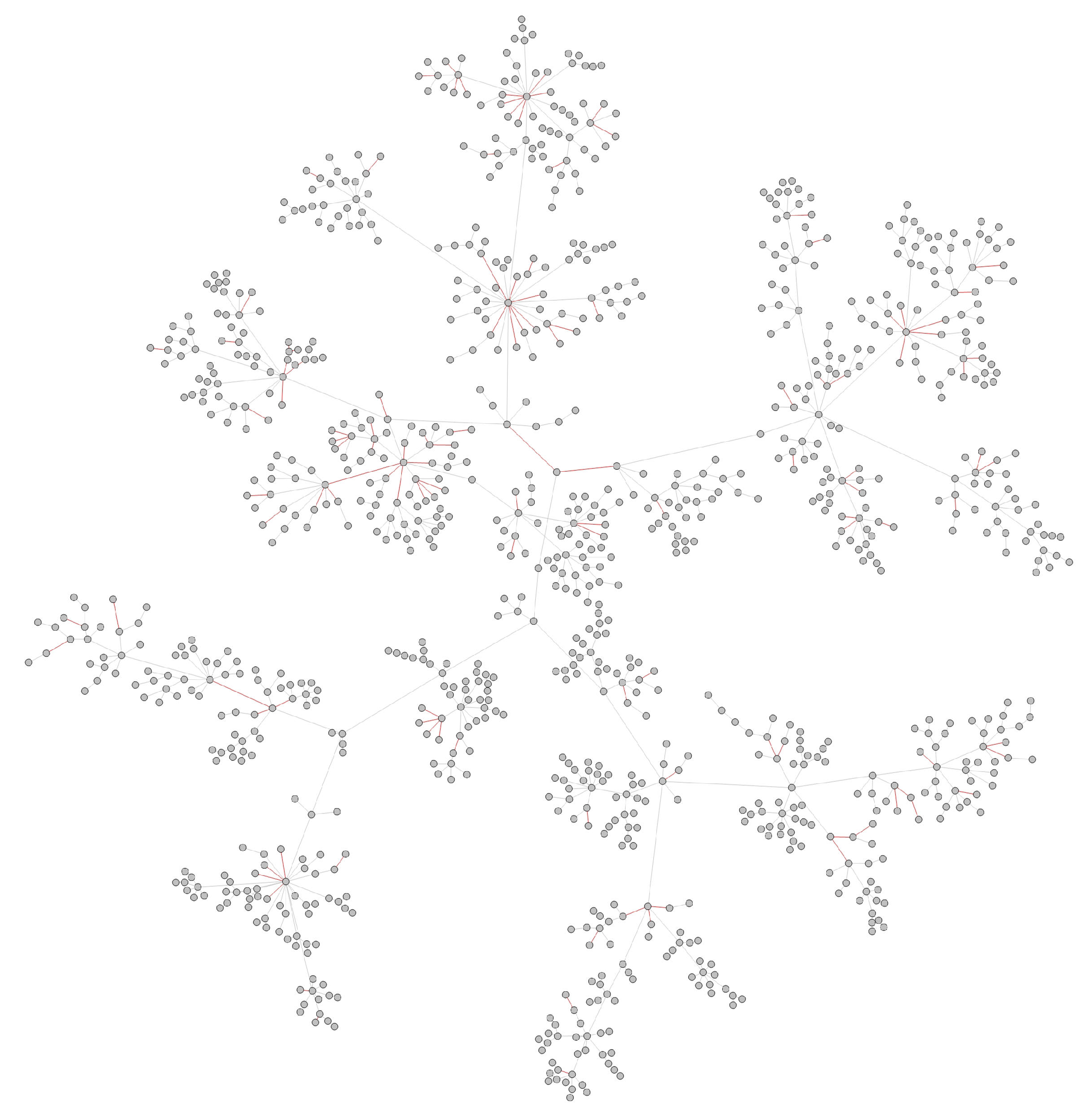
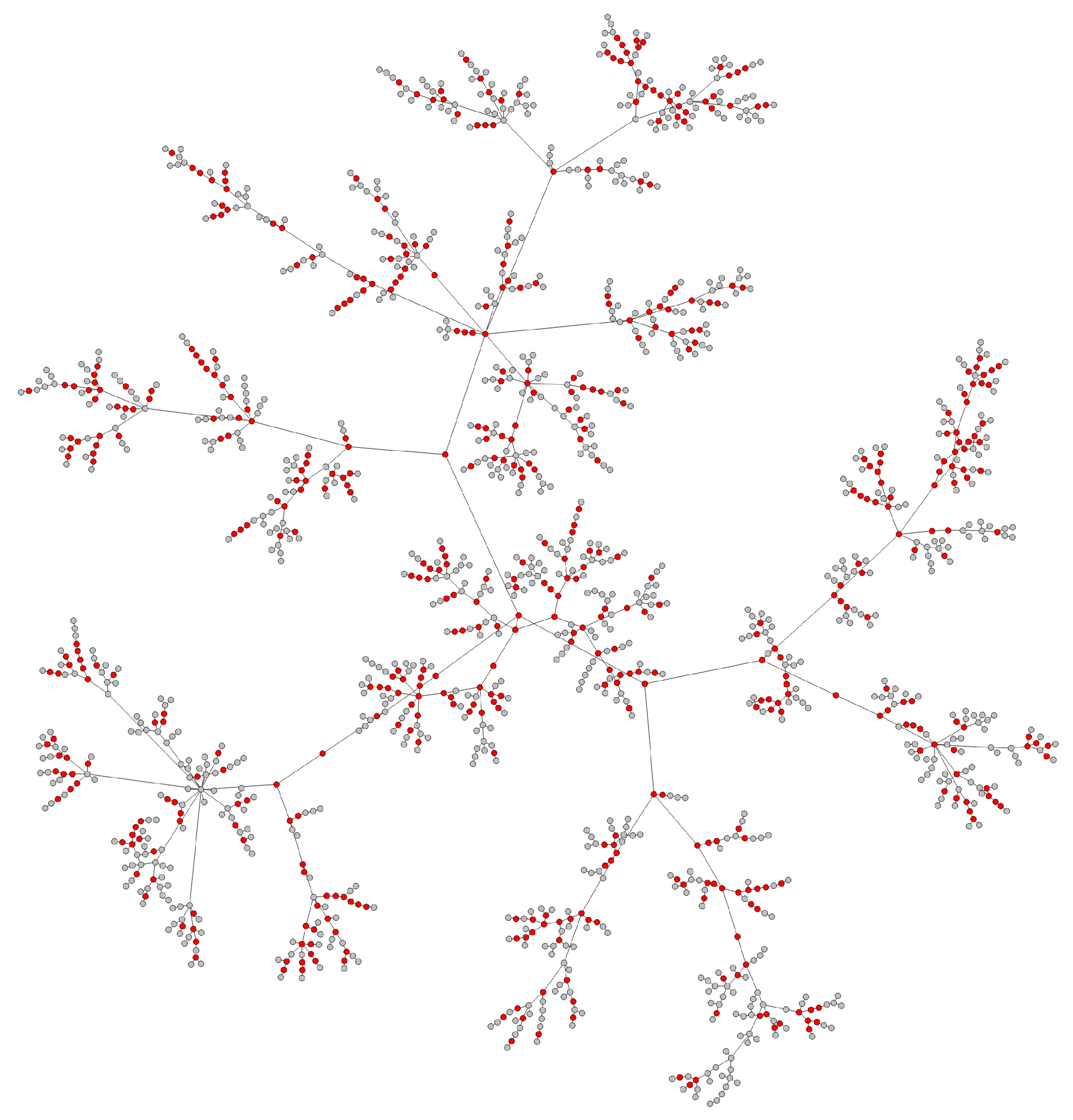
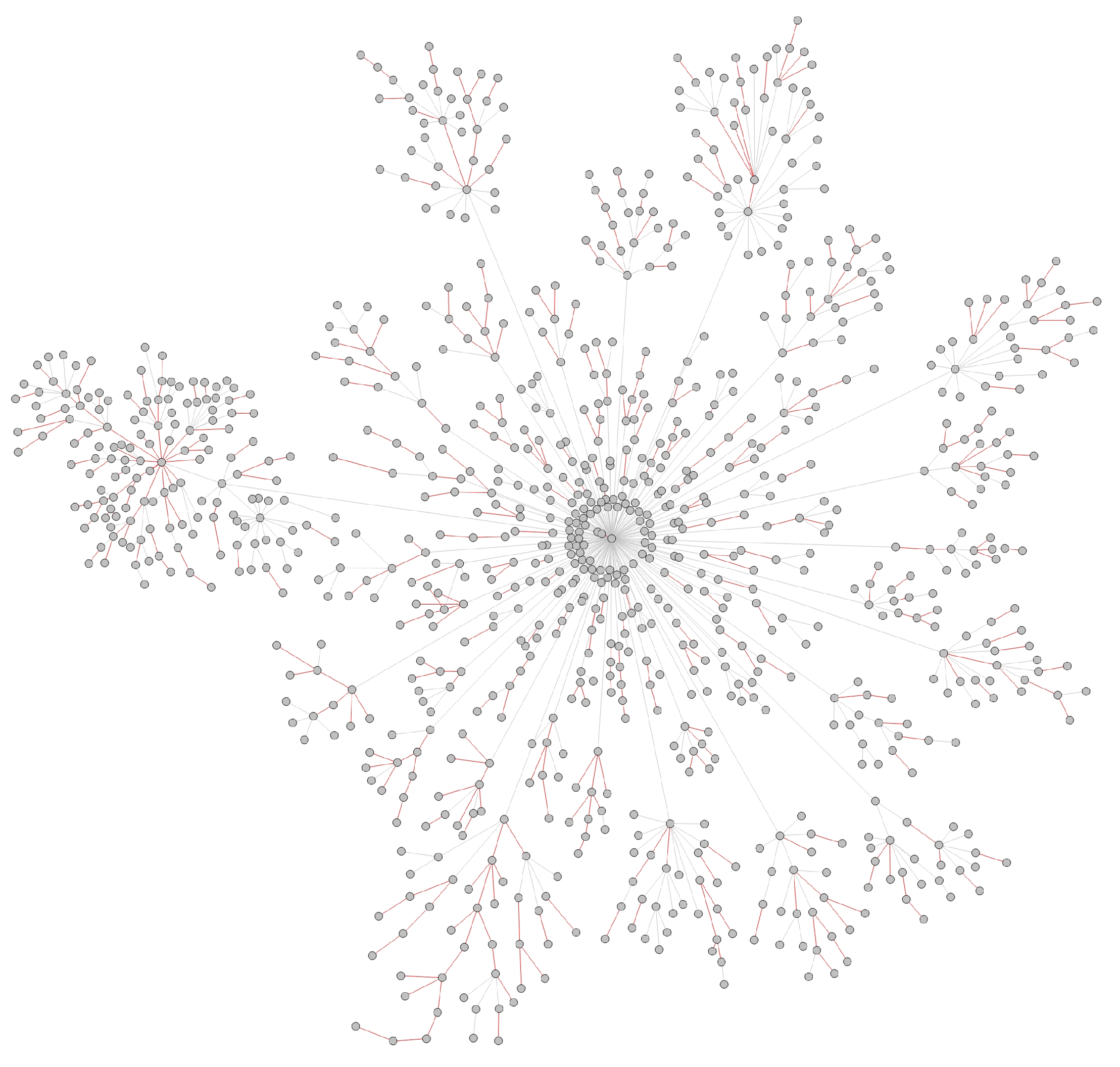
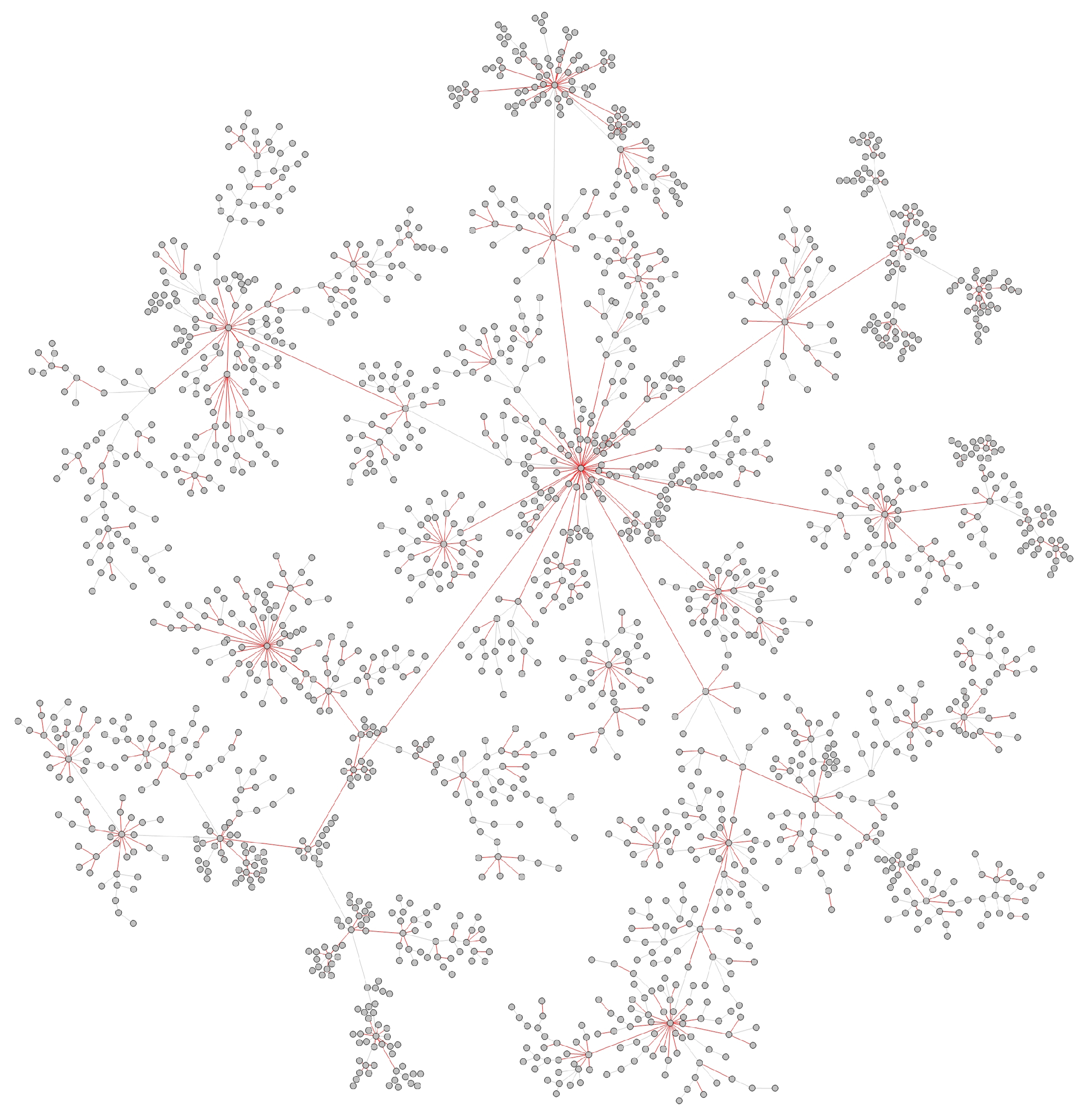
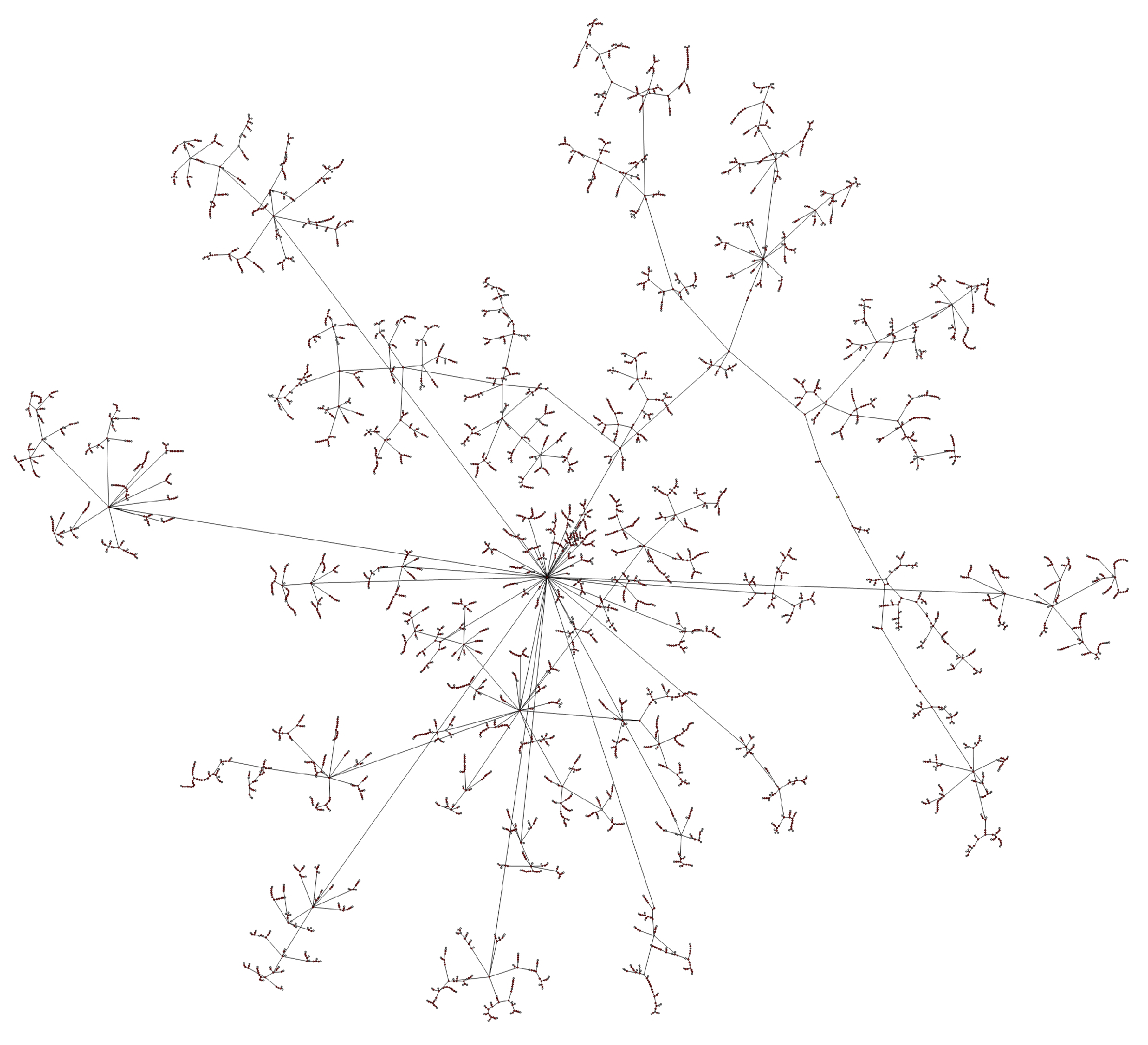
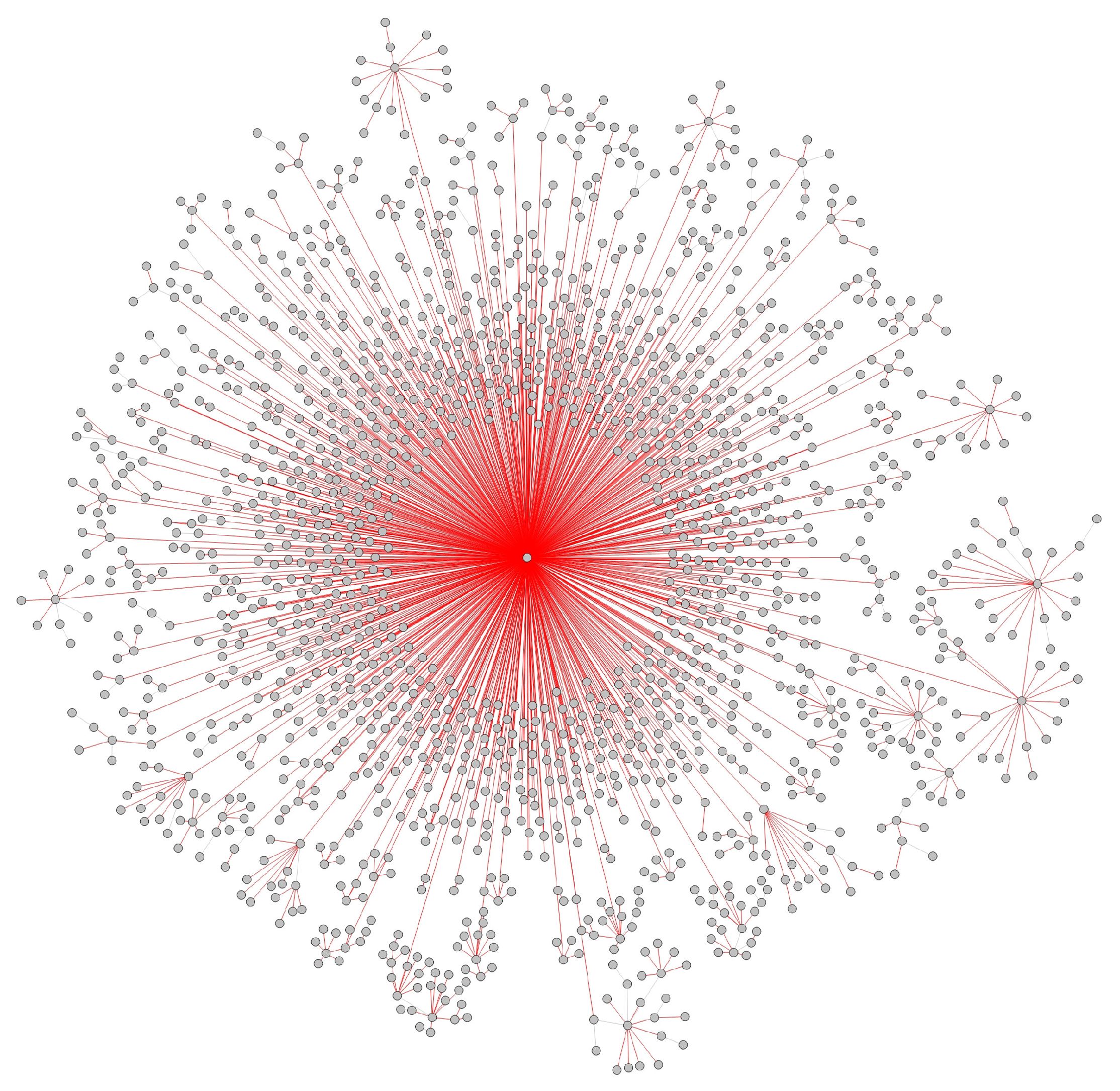
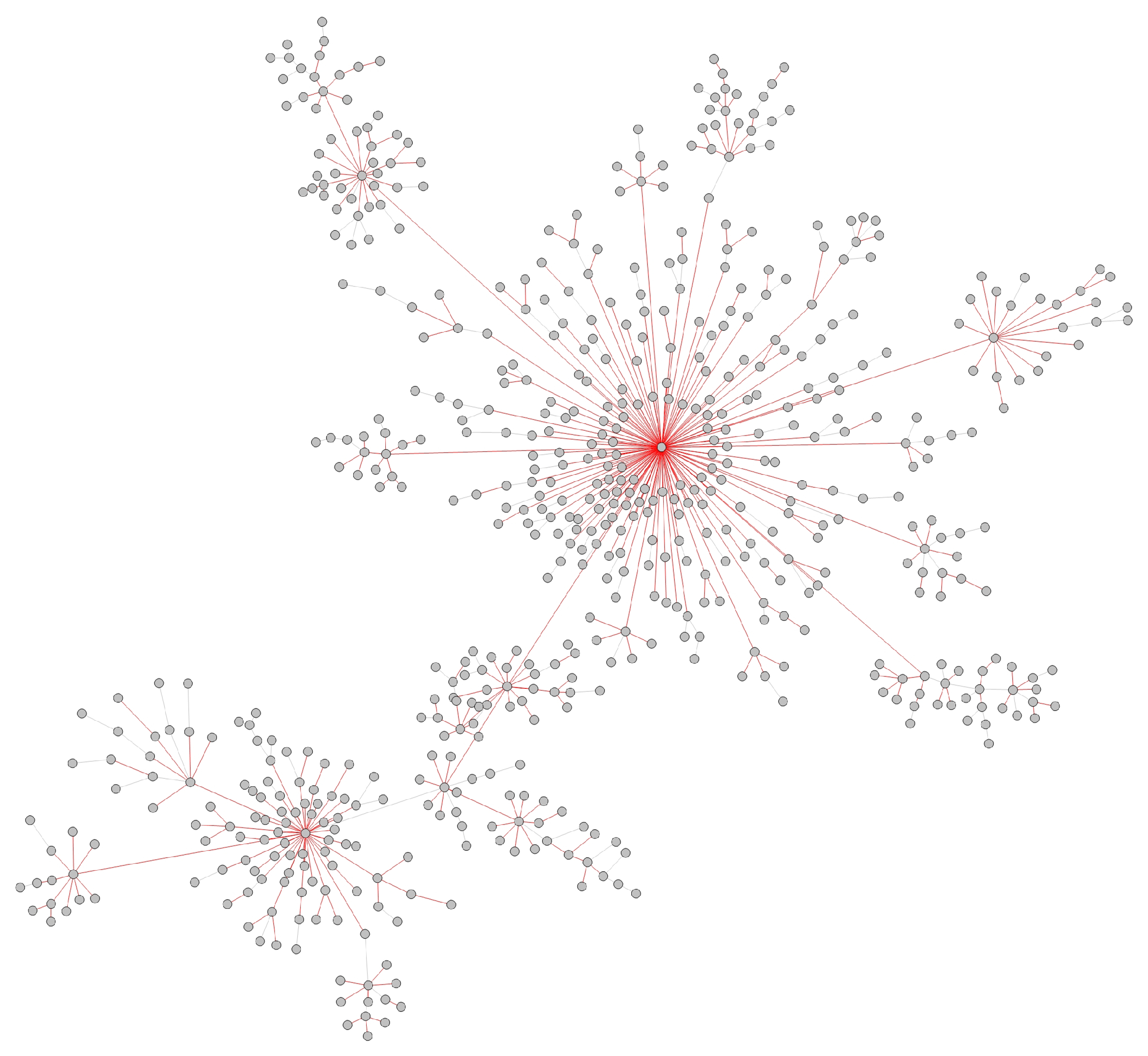
- Transmission networks of real dataIn this section, we show the transmission networks of different regions. Transmission networks were visualized using Gephi 0.9.2 which is a network analysis software [30]. Gephi’s built-in clustering algorithms Force Atlas 2 [31] is used to identify population clusters in the transmission network.In these figures, the red nodes represent inferred undetected spreaders in a transmission network. The red edges are abnormal edges with undetected nodes in them and are also used as test labels. Figure 7, Figure A1, Figure A3, Figure A5 and Figure A7 show the inferred transmission networks of Australia, Canada, Alberta, New York State and New Zealand respectively. Figure 8, Figure A2, Figure A4, Figure A6 and Figure A8 shows the observed transmission networks of Australia, Canada, Alberta, New York State and New Zealand respectively, which were also used as our test network. From these figures, we can see that the spread in different from region to region, and the distribution of the number of abnormal edges is different.








References
- Liu, Z.; Deardon, R.; Fu, Y.; Ferdous, T.; Cheng, Q. Estimating Parameters of Two-Level Individual-Level Models of the COVID-19 Epidemic Using Ensemble Learning Classifiers. Front. Phys. 2021, 8, 602722. [Google Scholar] [CrossRef]
- Kronbichler, A.; Kresse, D.; Yoon, S.; Lee, K.H.; Shin, J.I. Asymptomatic patients as a source of COVID-19 infections: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 98, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.Y.; Chen, L.; Wang, M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA J. Am. Med Assoc. 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, M.; Liang, W. The Dynamic COVID-Zero Strategy in China. Chin. Cent. Dis. Control Prev. 2022, 4, 74–75. [Google Scholar] [CrossRef]
- Burki, T. Dynamic zero COVID policy in the fight against COVID. Lancet Respir. Med. 2022, 10, e58–e59. [Google Scholar] [CrossRef]
- Popovich, K.J.; Snitkin, E.S. Whole Genome Sequencing—Implications for Infection Prevention and Outbreak Investigations. Curr. Infect. Dis. Rep. 2017, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Holmes, E.C.; Zhang, L.Q.; Robertson, P.; Cleland, A. The Molecular Epidemiology Of Human Immunodeficiency Virus Type 1 In Edinburgh. J. Infect. Dis. 1995, 171, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didelot, X.; Fraser, C.; Gardy, J.; Colijn, C. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks. Mol. Biol. Evol. 2018, 34, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2019, 4, 10–19. [Google Scholar] [CrossRef]
- Siobain, D. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar]
- Chen, C.; Yiwu, Z.; Wen, W.D. SARS-CoV-2: Apotential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, G.G. Natural Language Processing. Annu. Rev. Inf. Ence Technol. (ARIST) 2003, 37, 51–89. [Google Scholar] [CrossRef] [Green Version]
- Vanessa, I.; Jurtz, A.; Rosenberg, J.; Morten, N.; Jose, J. An introduction to deep learning on biological sequence data: Examples and solutions. Bioinformatics 2017, 33, 3685–3690. [Google Scholar]
- Velikovi, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Yuan, L.; Yuan, A.; Liu, M.; Hasan, S.A.; Hu, X. Integrating extra knowledge into word embedding models for biomedical NLP tasks. In Proceedings of the 2017 International Joint Conference on Neural Networks (IJCNN), Anchorage, AK, USA, 14–19 May 2017. [Google Scholar]
- Dong, Y.; Chawla, N.V.; Swami, A. Metapath2vec: Scalable Representation Learning for Heterogeneous Networks. In Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Halifax, NS, Canada, 13–17 August 2017. [Google Scholar]
- Han, J. Mining Heterogeneous Information Networks: Principles and Methodologies; Morgan & Claypool Publishers: San Rafael, CA, USA, 2012. [Google Scholar]
- Zhang, C.; Song, D.; Huang, C.; Swami, A.; Chawla, N.V. Heterogeneous Graph Neural Network. In Proceedings of the the 25th ACM SIGKDD International Conference, Anchorage, AK, USA, 4–8 August 2019. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-Supervised Classification with Graph Convolutional Networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Wang, D.; Peng, C.; Zhu, W. Structural Deep Network Embedding. In Proceedings of the Acm Sigkdd International Conference on Knowledge Discovery & Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Tharwat, A. Principal component analysis—A tutorial. Int. J. Appl. Pattern Recognit. 2016, 3, 197. [Google Scholar] [CrossRef]
- Gehring, J.; Auli, M.; Grangier, D.; Dauphin, Y.N. A Convolutional Encoder Model for Neural Machine Translation. arXiv 2016, arXiv:1611.02344. [Google Scholar]
- Japkowicz, N.; Shah, M. Evaluating Learning Algorithms: A Classification Perspective; Cambridge University Press: Cambridge, UK; Cambridge, MA, USA, 2011. [Google Scholar]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contributionto global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.; Perks, B.; Potemkin, M.; Gordon, P.; Long, Q. A Novel Computational Approach to Reconstruct SARS-CoV-2 Infection Dynamics through the Inference of Unsampled Sources of Infection; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2021. [Google Scholar]
- Mak, P.; Lang, K.; Marle, V. Evaluation of A Phylogenetic Pipeline to Examine Transmission Networks in A Canadian HIV Cohort. Microorganisms 2020, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andalibi, A.; Koizumi, N.; Li, M.H.; Siddique, A.B. Symptom and Age Homophilies in SARS-CoV-2 Transmission Networks during the Early Phase of the Pandemic in Japan. Biology 2021, 10, 499. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International ICWSM Conference, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Hu, Y. Efficient, High-Quality Force-Directed Graph Drawing. Math. J. 2006, 10, 37–71. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | AU | NZ | CA | AB | NY |
|---|---|---|---|---|---|
| Nodes | 1031 | 618 | 964 | 1847 | 1581 |
| Undetected nodes | 100 | 845 | 539 | 733 | 3031 |
| Abnormal edges | 113 | 439 | 526 | 828 | 1412 |
| Removed | Metric | RVTR | GCN | SDNE | AE | PCA | DIRECT |
|---|---|---|---|---|---|---|---|
| Precision | 0.71 ± 0.04 | 0.31 ± 0.02 | 0.33 ± 0.04 | 0.33 ± 0.04 | 0.62 ± 0.02 | 0.99 ± 0.00 | |
| AUC | 0.98 ± 0.01 | 0.53 ± 0.48 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.82 ± 0.23 | 0.98 ± 0.01 | |
| Precision | 0.91 ± 0.01 | 0.59 ± 0.05 | 0.59 ± 0.04 | 0.60 ± 0.03 | 0.88 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.97 ± 0.04 | 0.61 ± 0.41 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.87 ± 0.17 | 0.98 ± 0.00 | |
| Precision | 0.98 ± 0.01 | 0.89 ± 0.01 | 0.89 ± 0.01 | 0.89 ± 0.01 | 0.98 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.97 ± 0.04 | 0.57 ± 0.23 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.93 ± 0.05 | 0.98 ± 0.01 |
| Removed | Metric | RVTR | GCN | SDNE | AE | PCA | DIRECT |
|---|---|---|---|---|---|---|---|
| Precision | 0.68 ± 0.03 | 0.30 ± 0.02 | 0.30 ± 0.04 | 0.30 ± 0.03 | 0.60 ± 0.02 | 0.99 ± 0.00 | |
| AUC | 0.97 ± 0.04 | 0.52 ± 0.32 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.53 ± 0.24 | 0.97 ± 0.01 | |
| Precision | 0.85 ± 0.02 | 0.60 ± 0.02 | 0.62 ± 0.03 | 0.63 ± 0.03 | 0.87 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.95 ± 0.05 | 0.68 ± 0.44 | 0.55 ± 0.14 | 0.50 ± 0.00 | 0.63 ± 0.18 | 0.98 ± 0.00 | |
| Precision | 0.98 ± 0.01 | 0.91 ± 0.01 | 0.92 ± 0.01 | 0.92 ± 0.01 | 0.98 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.98 ± 0.02 | 0.45 ± 0.40 | 0.70 ± 0.24 | 0.50 ± 0.00 | 0.80 ± 0.11 | 0.98 ± 0.01 |
| Removed | Metric | RVTR | GCN | SDNE | AE | PCA | DIRECT |
|---|---|---|---|---|---|---|---|
| Precision | 0.69 ± 0.01 | 0.33 ± 0.03 | 0.34 ± 0.01 | 0.33 ± 0.02 | 0.58 ± 0.03 | 0.99 ± 0.00 | |
| AUC | 0.96 ± 0.09 | 0.50 ± 0.41 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.76 ± 0.18 | 0.96 ± 0.05 | |
| Precision | 0.89 ± 0.01 | 0.63 ± 0.02 | 0.61 ± 0.03 | 0.60 ± 0.02 | 0.84 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.98 ± 0.01 | 0.75 ± 0.38 | 0.49 ± 0.01 | 0.50 ± 0.00 | 0.76 ± 0.08 | 0.98 ± 0.01 | |
| Precision | 0.98 ± 0.01 | 0.92 ± 0.01 | 0.93 ± 0.01 | 0.93 ± 0.01 | 0.98 ± 0.01 | 0.99 ± 0.00 | |
| AUC | 0.99 ± 0.01 | 0.31 ± 0.05 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.83 ± 0.08 | 0.98 ± 0.00 |
| Removed | Model | Network | 1000 | 2000 | 3000 |
|---|---|---|---|---|---|
| 10% | RVTR-1K | Precision | 0.68 ± 0.03 | 0.62 ± 0.02 | 0.55 ± 0.02 |
| AUC | 0.99 ± 0.01 | 0.99 ± 0.01 | 0.97 ± 0.03 | ||
| RVTR-3K | Precision | 0.73 ± 0.04 | 0.70 ± 0.01 | 0.66 ± 0.02 | |
| AUC | 0.98 ± 0.03 | 0.99 ± 0.01 | 0.91 ± 0.16 | ||
| 20% | RVTR-1K | Precision | 0.89 ± 0.02 | 0.83 ± 0.02 | 0.80 ± 0.01 |
| AUC | 0.98 ± 0.02 | 0.99 ± 0.01 | 0.98 ± 0.01 | ||
| RVTR-3K | Precision | 0.91 ± 0.02 | 0.88 ± 0.01 | 0.87 ± 0.01 | |
| AUC | 0.98 ± 0.02 | 0.99 ± 0.01 | 0.96 ± 0.06 | ||
| 30% | RVTR-1K | Precision | 0.98 ± 0.01 | 0.97 ± 0.01 | 0.97 ± 0.01 |
| AUC | 0.98 ± 0.02 | 0.99 ± 0.00 | 0.99 ± 0.00 | ||
| RVTR-3K | Precision | 0.98 ± 0.01 | 0.98 ± 0.00 | 0.98 ± 0.00 | |
| AUC | 0.99 ± 0.01 | 0.98 ± 0.02 | 0.99 ± 0.00 |
| Removed | Metric | RVTR | GCN | SDNE | AE | PCA | DIRECT |
|---|---|---|---|---|---|---|---|
| Precision | 0.40 ± 0.07 | 0.24 ± 0.04 | 0.25 ± 0.05 | 0.23 ± 0.05 | 0.29 ± 0.03 | 0.26 ± 0.04 | |
| AUC | 0.99 ± 0.01 | 0.85 ± 0.28 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.45 ± 0.18 | 0.44 ± 0.22 | |
| Precision | 0.59 ± 0.03 | 0.46 ± 0.05 | 0.47 ± 0.04 | 0.46 ± 0.05 | 0.53 ± 0.03 | 0.49 ± 0.02 | |
| AUC | 0.96 ± 0.03 | 0.74 ± 0.37 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.41 ± 0.16 | 0.33 ± 0.09 | |
| Precision | 0.75 ± 0.04 | 0.69 ± 0.03 | 0.70 ± 0.02 | 0.68 ± 0.04 | 0.72 ± 0.01 | 0.71 ± 0.10 | |
| AUC | 0.95 ± 0.05 | 0.63 ± 0.43 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.48 ± 0.22 | 0.34 ± 0.10 |
| Dataset | RVTR | GCN | SDNE | AE | PCA | Direct |
|---|---|---|---|---|---|---|
| NZ | 0.7904 | 0.6651 | 0.8064 | 0.6720 | 0.6720 | 0.8009 |
| CA | 0.6654 | 0.5000 | 0.5627 | 0.5627 | 0.6103 | 0.5760 |
| AB | 0.5399 | 0.3599 | 0.4348 | 0.4348 | 0.4855 | 0.4469 |
| NY | 0.9363 | 0.8916 | 0.9186 | 0.8916 | 0.9143 | 0.9186 |
| Removed | Metric | |||
|---|---|---|---|---|
| 10% | Precision | 0.32 ± 0.06 | 0.73 ± 0.04 | 0.72 ± 0.05 |
| AUC | 0.73 ± 0.17 | 0.98 ± 0.03 | 0.80 ± 0.29 | |
| 20% | Precision | 0.54 ± 0.06 | 0.91 ± 0.02 | 0.98 ± 0.01 |
| AUC | 0.81 ± 0.14 | 0.98 ± 0.02 | 0.67 ± 0.24 | |
| 30% | Precision | 0.74 ± 0.05 | 0.98 ± 0.01 | 0.99 ± 0.01 |
| AUC | 0.80 ± 0.19 | 0.99 ± 0.01 | 0.66 ± 0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Ma, Y.; Cheng, Q.; Liu, Z. Finding Asymptomatic Spreaders in a COVID-19 Transmission Network by Graph Attention Networks. Viruses 2022, 14, 1659. https://doi.org/10.3390/v14081659
Liu Z, Ma Y, Cheng Q, Liu Z. Finding Asymptomatic Spreaders in a COVID-19 Transmission Network by Graph Attention Networks. Viruses. 2022; 14(8):1659. https://doi.org/10.3390/v14081659
Chicago/Turabian StyleLiu, Zeyi, Yang Ma, Qing Cheng, and Zhong Liu. 2022. "Finding Asymptomatic Spreaders in a COVID-19 Transmission Network by Graph Attention Networks" Viruses 14, no. 8: 1659. https://doi.org/10.3390/v14081659
APA StyleLiu, Z., Ma, Y., Cheng, Q., & Liu, Z. (2022). Finding Asymptomatic Spreaders in a COVID-19 Transmission Network by Graph Attention Networks. Viruses, 14(8), 1659. https://doi.org/10.3390/v14081659