1. Introduction
Since emerging in Wuhan, China, in late 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused over 6.9 million confirmed deaths worldwide (
https://covid19.who.int/, accessed 28 August 2023) to August 2023 and severely affected the world economy [
1]. Once the World Health Organization (WHO) declared SARS-CoV-2 a pandemic in March 2020, researchers strove for the quick development of novel vaccines and drugs to combat the spread of coronavirus disease 2019 (COVID-19). Because of these urgent circumstances, vaccination platforms that allow rapid engineering and simultaneously deliver a robust immune response, such as the mRNA or adenoviral vaccination platforms, asserted themselves against classical vaccination regimes such as live-attenuated vaccines. Adenoviral and mRNA vaccination platforms allow immediate reaction to new pathogens or strains by altering the sequence of the to-be-delivered nucleic acid. This diversity in vaccine regimens was one major tool controlling the spreading of the pandemic. In particular, adenoviral vaccines offer several advantages as an emergency outbreak vaccination platform, such as high target cell transduction and gene transfer efficacy, as well as triggering robust humoral and cellular immune responses and prolonged stability at room temperature for storage [
2]. The design of such adenoviral vectors for the expression of a specific transgene is not trivial, as the expression of particular transgenes may affect viral propagation and may thus require a protein-dependent design of the expression cassette. For efficient vaccine production, optimal vector design relies on the perfect balance between transgene expression and vector functionality.
Here, we want to share our knowledge gained by generating a series of human adenovirus type 5 (HAdV-C5) vector-based vaccine candidates. While a majority of adenoviral vaccines that gained conditional market approval are based on tetracycline-controlled gene expression systems [
3,
4], our vector design relies on constitutive protein expression in transduced target cells. This way, effects of transgene expression, such as antigen-related toxicity on human cells, can be observed, enabling early estimation of patient safety following vaccination. SARS-CoV-2 is an ssRNA virus, and the spike protein is encoded by RNA. Therefore, SARS-CoV-2 RNA is never confronted with the RNA-processing machinery in the nucleus but is immediately translated in the cytoplasm. To clone the spike gene into an adenoviral genome, a DNA sequence originating from the RNA sequence has to be generated. Consequently, this DNA (of a sequence never meant for being localized in a nucleus) needs to be transcribed and processed before leaving the nucleus. As such, transferring the sequence of a genuine RNA gene to a DNA vector bears the risk of aberrant splicing and, as a consequence, defective transgene products. Therefore, obtaining a substantial understanding regarding the contextualizing of genes from RNA viruses into a DNA background is an essential basis for future developments of adenoviral vaccines.
With this study, we want to share our knowledge gained while generating our own set of adenovirus type 5 vector-based vaccine candidates harboring a CMV (cytomegalovirus)promoter-driven spike expression cassette located within the HAdV-C5 E1-region. We report that physical titers, viral propagation and spike protein expression were affected based on different expression cassette designs. While mRNA splicing of transcripts was seemingly not affected, fusogenicity of ACE2 (angiotensin-converting enzyme)-overexpressing cells was enhanced, dependent on the design of adenoviral vectors. We think that these insights into our struggles with vector production will be useful for future vaccine development.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
HEK293 (ATCC®, Manassas, VA, USA, CRL-1573™) and A549 (ATCC®, Manassas, VA, USA, CLL-185™) cells were cultured in MEM Eagle (PAN Biotech™, Aidenbach, Germany, P04-08500) supplemented with 10% FCS (PAN Biotech™, Aidenbach, Germany, P40-37500) and 1% penicillin–streptomycin (PAN Biotech™, Aidenbach, Germany, P06-07050). U2OS-GFP-ACE2 cells were generated following transduction and selection with a lentivirus encoding the SARS-CoV-2 receptor angiotensin-converting enzyme 2 (ACE2, addgene, Watertown, MA, USA, #145839) and maintained under selection of 5 µg/mL puromycin (Merck KGaA, Darmstadt, Germany, P7255-25MG). U2OS, as well as KM-12 (Cellosaurus, CVCL_1331), Huh-7 (Cellosaurus, CVCL_0336) and SH-SY5Y (ATCC®, Manassas, VA, USA, CRL-2266™) were maintained in DMEM (PAN Biotech™, Aidenbach, Germany, P04-03590) supplemented with 10% FCS and 1% penicillin–streptomycin (PAN Biotech™, Aidenbach, Germany, P06-07050). Cells were passaged twice per week with 0.05% trypsin–0.02% EDTA (PAN Biotech™, Aidenbach, Germany, P10-0231SP).
2.2. Vector Construction
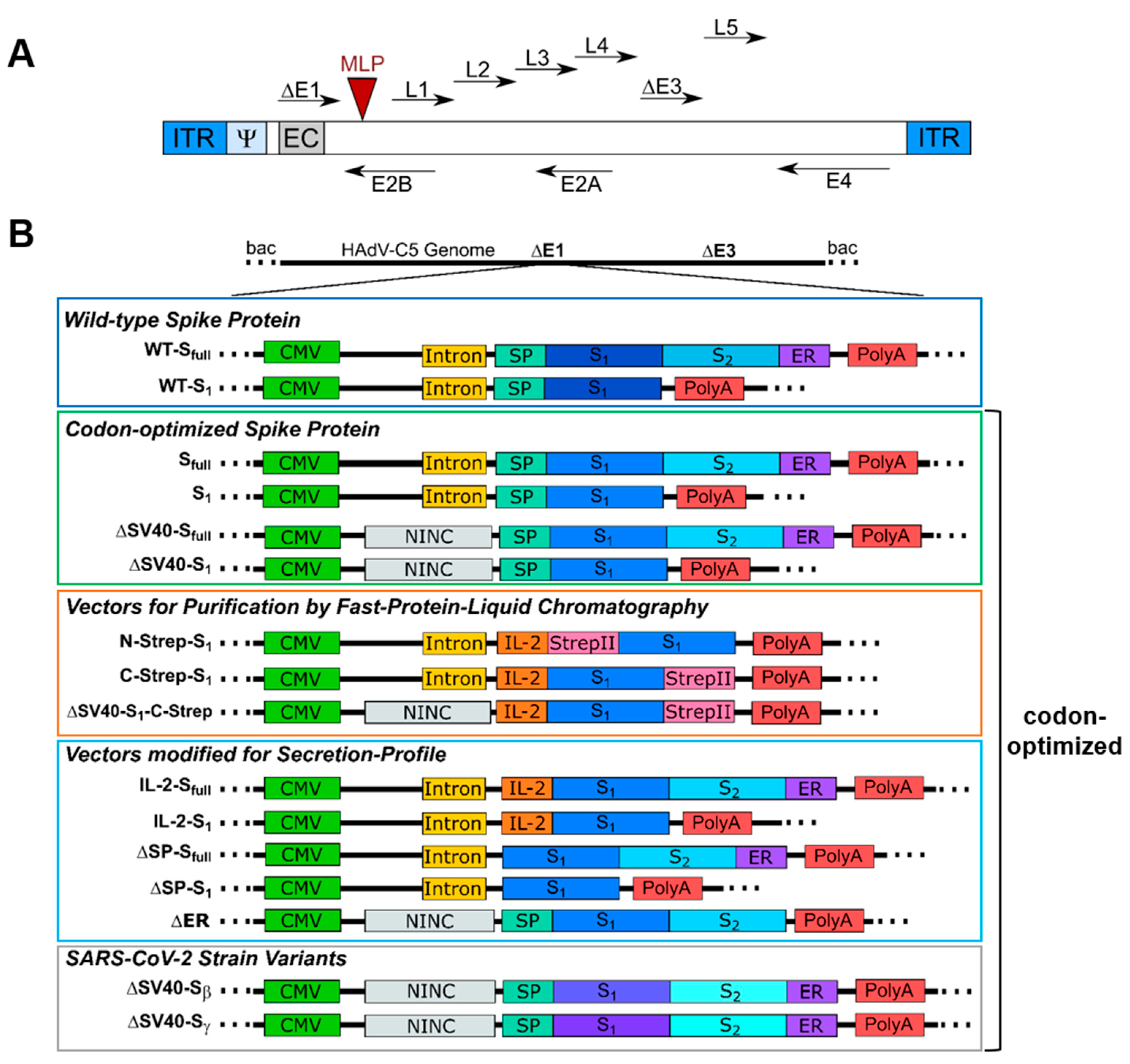
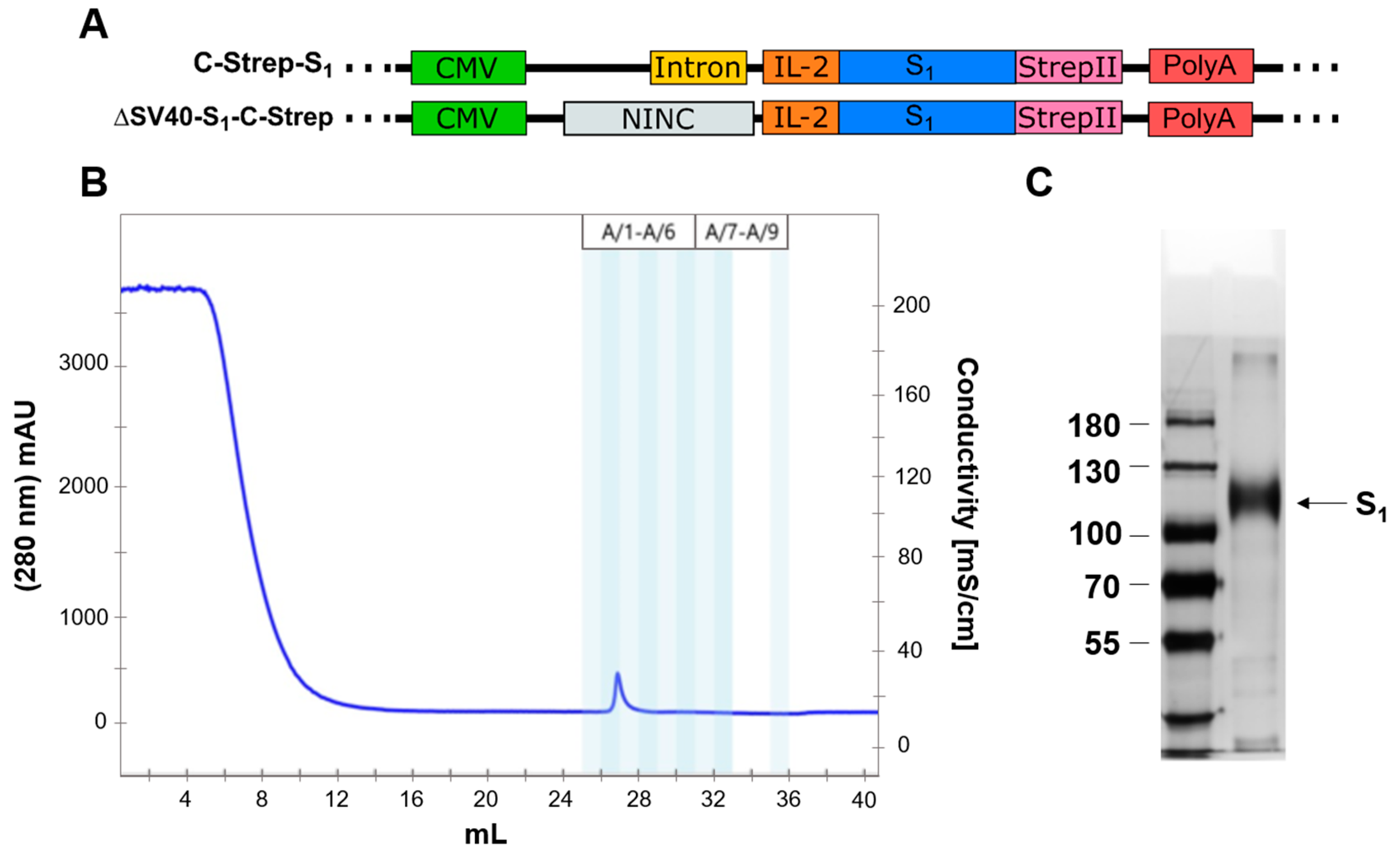
For this study, each E1/E3-deleted vector of human adenovirus type 5 (AY339865; bp 1–440, bp 3522–28131, bp 30814–35934) was generated using the Counter-Selection BAC Modification Kit (GeneBridges GmbH, Heidelberg, Germany, K002), which is based on pRed/ET recombination. Briefly, using in vivo homologous recombination in
Escherichia coli (
E. coli) DH10β, a counter-selection rpsL-neo cassette with flanking homology arms was introduced in an E1-located expression cassette consisting of a CMV promoter and an SV40 intron. The rpsL-neo cassette was either replaced by the Wuhan isolate (GenScript Biotech Corporation, Piscataway Township, NJ, USA, NC_045512.2) or codon-optimized (pUC57-2019-nCOV-S, GenScript Biotech Corporation, Piscataway Township, NJ, USA) DNA sequence of the full-length spike protein or only the S
1 domain. Furthermore, the SV40 intron, the N-terminally located signal peptide (SP) of the spike protein, or the endoplasmic reticulum (ER) retention signal of the spike protein was deleted by placement of the rpsL-neo cassette and subsequently replaced by a non-coding non-intronic sequence in the case of the intron or by the IL-2 signal peptide in the case of the spike signal peptide (for reference, see
Section 3.1).
2.3. Adenovirus Vector Purification
Before transfection of cells, adenoviral vectors were liberated from bacterial bacmid backbone by restriction digestion with SwaI (New England Biolabs GmbH, Ipswich, MA, USA, R0604S). In short, 2.5 µg of DNA was incubated with 3 U SwaI in supplied buffer at 25 °C overnight. HEK293 cells were seeded at a density of 1 × 10
5 cells/well in a 24-well plate and cultivated overnight. The next day, 500 ng DNA was premixed with a 150 mM NaCl solution to a volume of 25 µL, and 6 µL of a 7.5 mM linear polyethyleneimine (PEI) (22 kDa) solution was mixed with a 150 mM NaCl solution to a volume of 25 µL. PEI mixture was added to the DNA solution and incubated at room temperature (RT) for 10 min. Subsequently, the complete PEI-DNA mixture was added to one well of the 24-well plate. Cells were harvested once cytopathic effects became visible after roughly 12 to 14 days. Lysates were used for cycles of reinfections and vector particles were finally purified using CsCl density gradient centrifugation. Two purification steps using discontinuous CsCl gradients (1.27 g/cm
3 and 1.41 g/cm
3) were performed, and vector bands were collected after centrifugation at 4 °C and 13.200 rpm for 2 h. Preparations were desalted using PD10 columns (Cytiva, Marlborough, MA, USA, 17085101). For the determination of physical titers, 20 µL of vector preparation was mixed with 0.1% SDS and 79 µL buffer and incubated at 56 °C for 10 min. Extinction at 260 nm was measured using UV spectroscopy, and titers were calculated utilizing the following formula:
where vp = virus particles, E
260 = extinction at 260 nm, and DF = dilution factor
2.4. Polymerase Chain Reaction (PCR)
DNA fragments for homologous recombination were generated using Q5
® high-fidelity polymerase (New England Biolabs GmbH, Ipswich, MA, USA, M0491L) with primers including 50 bp homology arms to the insertion site. Thus, 10 ng DNA template were mixed with 0.04 U/µL polymerase, 0.2 mM dNTPs, 1 µM primers forward and reverse (
Table S1), as well as the appropriate buffer. The following program was used: cycle 1—98 °C, 120 s; cycles 2–27—98 °C, 30 s, then 67 °C, 30 s, followed by 72 °C, 120 s; cycle 28: 72 °C, 180 s. Colony PCR was performed to verify the replacement of rpsL-neo cassette by target sequence. As such, picked colonies were cultured in 100 µL LB medium supplemented with respective antibiotics and incubated at 37 °C for 3 h. Thereafter, 3 µL of bacterial culture was added to a mixture of 1 µM forward and reverse primers (forward: 5′ GCTCGTTTAGTGAACCGTCAGA 3′, reverse: 5′ GAGGCCGAGTTTGTCAGAAAGC 3′), 0.2 mM dNTPs, 0.05 U/µL GoTaq
® polymerase (Promega Corporation, Madison, WI, USA, M3001) in appropriate buffer. PCR reaction was performed with the following program: cycle 1—95 °C, 180 s; cycles 2–31—95 °C, 45 s, then 58 °C, 45 s, followed by 74 °C, 120 s; cycle 32—74 °C, 500 s. For the analysis of mRNA splicing of transcripts, a 3 µL DNA sample was mixed with 0.2 mM dNTPs, 0.05 U/µL GoTaq
® polymerase (Promega Corporation, Madison, WI, USA, M3001), and 1 µM forward and reverse primers (
Table S2) in appropriate buffer. PCR cycles were: cycle 1—95 °C, 180 s; cycles 2–31—95 °C, 70 s, then 59 °C, 70 s, followed by 74 °C, 300 s; cycle 32—74 °C, 600 s.
2.5. Sodium Dodecyl Polyacrylamide Gel Electrophoresis (SDS-PAGE)
For adjustment of titer from viral preparations, 5 × 10
9 viral particles (vp), as calculated by physical titers, were diluted in Ad buffer (150 mM NaCl, 50 mM HEPES, pH 8.0) to a final volume of 20 µL. SDS loading buffer (5 µL) was added, and samples were boiled for 5 min at 95 °C. Samples were loaded onto an 8% polyacrylamide gel, which after electrophoresis was stained using silver staining (see
Section 2.6).
Spike protein expression by viral vectors was analyzed from either supernatant or cell pellet. Thus, 100 µL of supernatant after infection of HEK293 or transduction of A549 or U2OS cells was transferred to a reaction tube. Protein extraction from cells was performed according to a modified protocol of the original by K. K. Wang [
5]. Cells were washed once with 1 mL DPBS (PAN Biotech™, Aidenbach, Germany, P04-36500), then treated with 300 µL RIPA-SDS buffer with protease-inhibitor cocktail (Merck KGaA, Darmstadt, Germany, 11836153001) and incubated at room temperature for 10 min. After addition of 100 µL 100% trichloroacetic acid (TCA), the supernatant was transferred to a reaction tube. The pellet after centrifugation (RT, 5000×
g, 5 min) was washed with 1 mL 2.5% TCA and again centrifuged (RT, 5000×
g, 5 min). Tris base (20 µL, 3 M) was added to the pellet after centrifugation, and samples were then incubated at room temperature for 30 min. Finally, samples were mixed with 20 µL ddH
2O and 10 µL of 5× SDS sample buffer, then incubated at 95 °C for 10 min. Samples were loaded onto an 8% polyacrylamide gel, and separated proteins, after electrophoresis, were thereafter transferred to a nitrocellulose membrane (see
Section 2.9).
2.6. Silver Staining of Polyacrylamide Gel
Silver staining of polyacrylamide gels was performed according to H. Blum et al. [
6].
Briefly, polyacrylamide gel after electrophoresis was transferred into a fixation solution (50% (v/v) methanol, 12% (v/v) acetic acid, 13.6 mM formaldehyde) and incubated at room temperature for 30 min. The gel was then rinsed with a washing buffer (50% ethanol) for 15 min and then treated with pretreatment buffer (0.8 mM sodium thiosulfate) for 1 min. After washing thrice for 20 s with ddH2O, the gel was incubated in impregnation buffer (0.2% (w/v) silver nitrate, 13.6 mM formaldehyde) for 20 min at room temperature. Gel was subsequently developed in developer solution (6% (w/v) sodium carbonate, 16 µM sodium thiosulfate, 13.6 mM formaldehyde) after washing twice with ddH2O for 20 s. The reaction was stopped by treatment with stopping buffer (50% (v/v) methanol, 12% (v/v) acetic acid) after washing with ddH2O twice for 2 min.
2.7. Infection of HEK293 Cells with Adenoviral Vectors
For real-time quantitative PCR (RT-qPCR) analysis, 3 × 104 HEK293 cells/well were seeded in a 96-well plate and cultured overnight. The next day, cells were infected with spike protein expressing HAdV-C5 vectors with a multiplicity of infection (MOI) of 200 according to adjusted titers and incubated for 48 h at 37 °C, 5% CO2. For the analysis of mRNA splicing, 2 × 106 HEK293 were seeded in 6 cm cell culture dishes and cultured overnight. The following day, cells were infected with an MOI of 300 and incubated at 37 °C and 5% CO2 for 48 h.
2.8. Transduction of A549 or U2OS Cells with Adenoviral Vectors
For the analysis of mRNA splicing, 2 × 106 A549 cells were seeded in 6 cm cell culture dishes a day prior to transduction. Cells were then transduced with an MOI of 300 and incubated at 37 °C and 5% CO2 for 48 h.
For Western blot analysis of spike protein expression, 2 × 105 U2OS cells/well were seeded in a 24-well plate and cultured overnight. Cells were transduced with spike protein encoding viral vectors using an MOI of 300 according to adjusted titers and incubated for 48 h at 37 °C, 5% CO2. Furthermore, to enhance understanding of spike protein expression, 2 × 105 A549 cells/well in a 24-well plate were transduced with adenoviral vectors with an MOI of 1000 according to adjusted titers and incubated for 48 h at 37 °C, 5% CO2.
2.9. Fast Protein Liquid Chromatography (FPLC) for Spike Protein Purification
In order to purify glycosylated S1 protein from media after secretion, at least 10 × 15 cm cell culture dishes of A549 cells were transduced with appropriate vector using an MOI of 3000. The next day, cells were washed thrice with 15 mL of warm medium and DPBS. Cells were then treated with 25 mL EX-CELL® (Merck KGaA, Darmstadt, Germany, 14571C-1000ML) medium and incubated at 37 °C, 5% CO2 for an additional 48 h until the medium was harvested. The supernatant was mixed with cOmplete™ protease-inhibitor cocktail (Merck KGaA, Darmstadt, Germany, 11697498001), then centrifuged for 8 min at 300× g. The supernatant was transferred into centrifuge tubes, and 5 µL [0.015 U/µL] of an avidin solution (IBA Lifesciences GmbH, Göttingen, Germany, 2-0204-050) was added. After incubation at room temperature for 20 min, tubes were centrifuged at 18.000× g and 4 °C for 30 min.
For protein purification, an NGC Quest 10 chromatography system (Bio-Rad Laboratories GmbH, Hercules, CA, USA, #7880001) was used. Subsequently, the supernatant was loaded onto a StrepTactin™ column (Cytiva, Marlborough, MA, USA, 29048653). Purification of S1 protein from the column was performed according to the manufacturer’s instructions. Product fractions after purification were concentrated to a total volume of 100 µL using Amicon® ultra centrifugal units (Merck KGaA, Darmstadt, Germany, UFC503096) according to the product’s manual.
2.10. Western Blot
After subjecting protein samples to SDS-PAGE, proteins were transferred to a nitrocellulose membrane (Cytiva, Marlborough, MA, USA, 10600003). Membranes were blocked with 5% bovine serum albumin in TBS-T (Tris-buffered saline, 100 mM Tris and 150 mM NaCl, with 0.05% Tween-20) overnight at 4 °C. For this study, either the rabbit-α-spike antibody (Sino Biological, Beijing, China, 40592-T62; 1:1000 in 5% BSA/TBS-T) or human serum after vaccination (1:20 in 5% BSA/TBS-T) was used as primary antibody. Membranes were incubated with primary antibody for 1 h at room temperature, then washed five times for at least 5 min with TBS-T. Secondary antibodies used were either goat-α-rabbit-IRDye800CW antibody (LI-COR Biosciences, Lincoln, NE, USA, 926-32211; 1:15.000 in 5% BSA/TBS-T) or goat-α-human-IRDye680RD antibody (LI-COR Biosciences, Lincoln, NE, USA, 926-68078; 1:15.000 in 5% BSA/TBS-T). Membranes were incubated with the appropriate secondary antibody for 45 min at room temperature. After subsequent washing steps with TBS-T, membranes were documented with the LI-COR Odyssey® CLx imaging system.
2.11. Isolation of Viral DNA for Quantitative PCR Analysis
Viral DNA from supernatant 48 h after infection of HEK293 cells was used for analyzing viral particle production using qPCR. Thus, the 96-well plate was subjected to three cycles of freezing and thawing to liberate viral particles from still-adherent cells. Supernatant after centrifugation (RT, 300× g, 5 min) was transferred to 1.5 mL reaction tube. Viral DNA was then isolated using a Quick-DNA Miniprep Plus Kit (Zymo Research Europe GmbH, Freiburg, Germany, D4069) following the instructions of the manual, and sample concentration was determined by UV spectroscopy. For qPCR, DNA samples were diluted to a concentration of 7 ng/µL and stored at 4 °C until measurement.
2.12. Isolation of Total RNA and cDNA Synthesis
Total RNA was isolated using TRIzol™ (Thermo Fisher Scientific, Waltham, MA, USA, 15596026) following the instructions of the provided manual. Cells seeded in 6 cm cell culture dishes were harvested in 1 mL TRIzol™. RNA samples were stored at −80 °C or immediately reverse-transcribed into cDNA using the PrimeScript™ first strand cDNA synthesis kit (Takara Bio Inc., Kusatsu, Japan, 6110A) following the manufacturer’s instruction manual. Prior to cDNA synthesis, 10 µg RNA was digested with 10 U DNAse I (Thermo Fisher Scientific, Waltham, MA, USA, EN0521) in MgCl2 buffer for 3 h at 37 °C. cDNA was diluted with 60 µL ddH2O for the analysis of mRNA splicing by PCR and stored at −20 °C until further use.
2.13. Quantitative PCR Analysis
Viral production was quantified by qPCR analysis. As such, 1.5 µL of isolated viral DNA was added to 10 µL TB Green Advantage Premix (Takara Bio Inc., Kusatsu, Japan, 639676), 0.2 µM of appropriate forward and reverse primer (E4 forward: 5′ TAGACGATCCCTACTGTACG 3′, E4 reverse: 5′ GGAAATATGACTACG TCCGG 3′, PLAT forward: 5′ AGGGCTGGAGAGAAAACCTC 3′, PLAT reverse: 5′ TTCCTTCACTGGCTCAGCTT 3′) and adjusted with ddH2O to a final volume of 20 µL. The following program was used for template amplification: cycle 1—95 °C, 1 min; cycles 2–46—95 °C, 10 s followed by 60 °C, 25 s. The E4 copy number of each sample determined by qPCR was normalized to PLAT (plasminogen activator, tissue type) concentration.
2.14. Bright-Field Microscopic Imaging
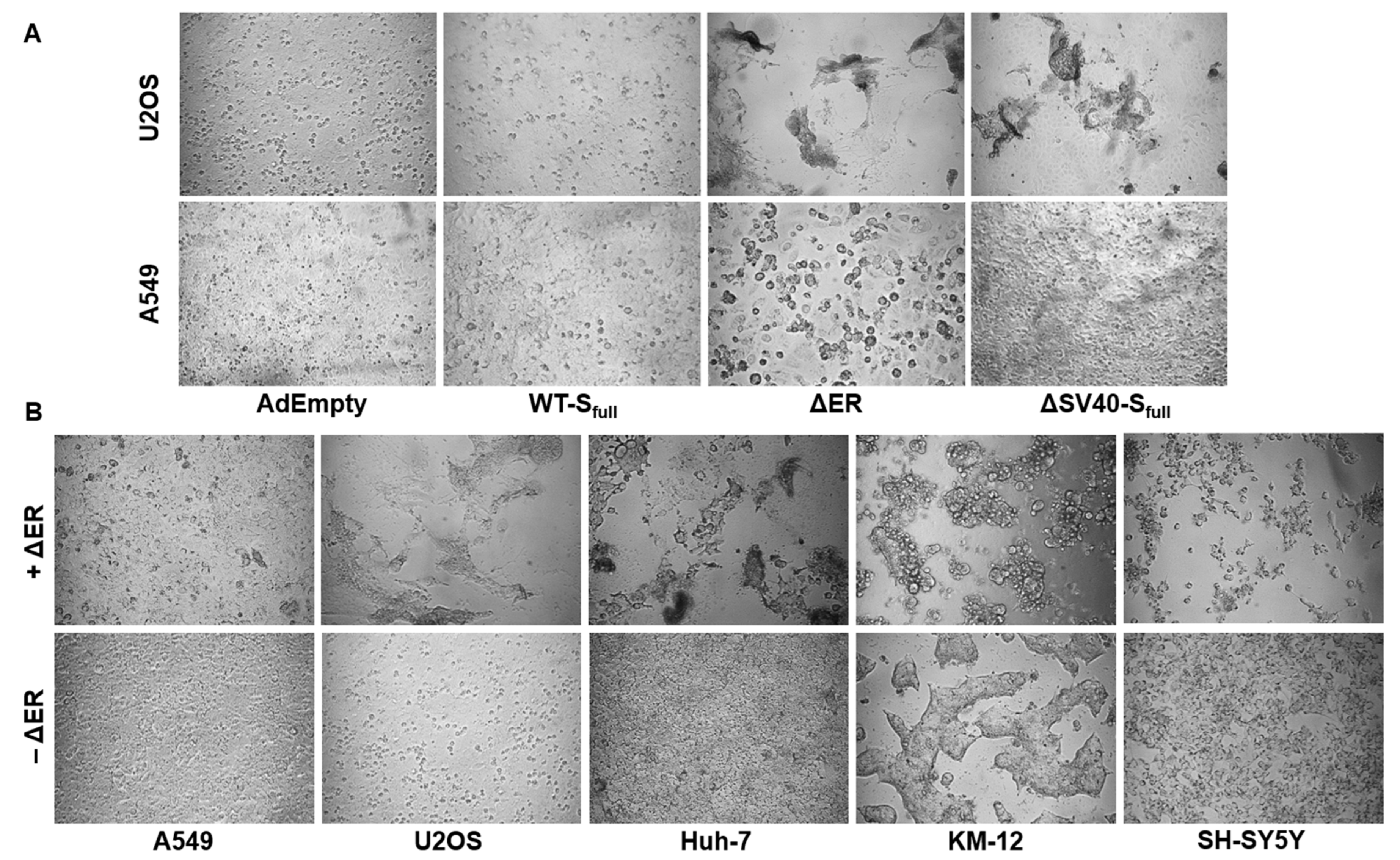
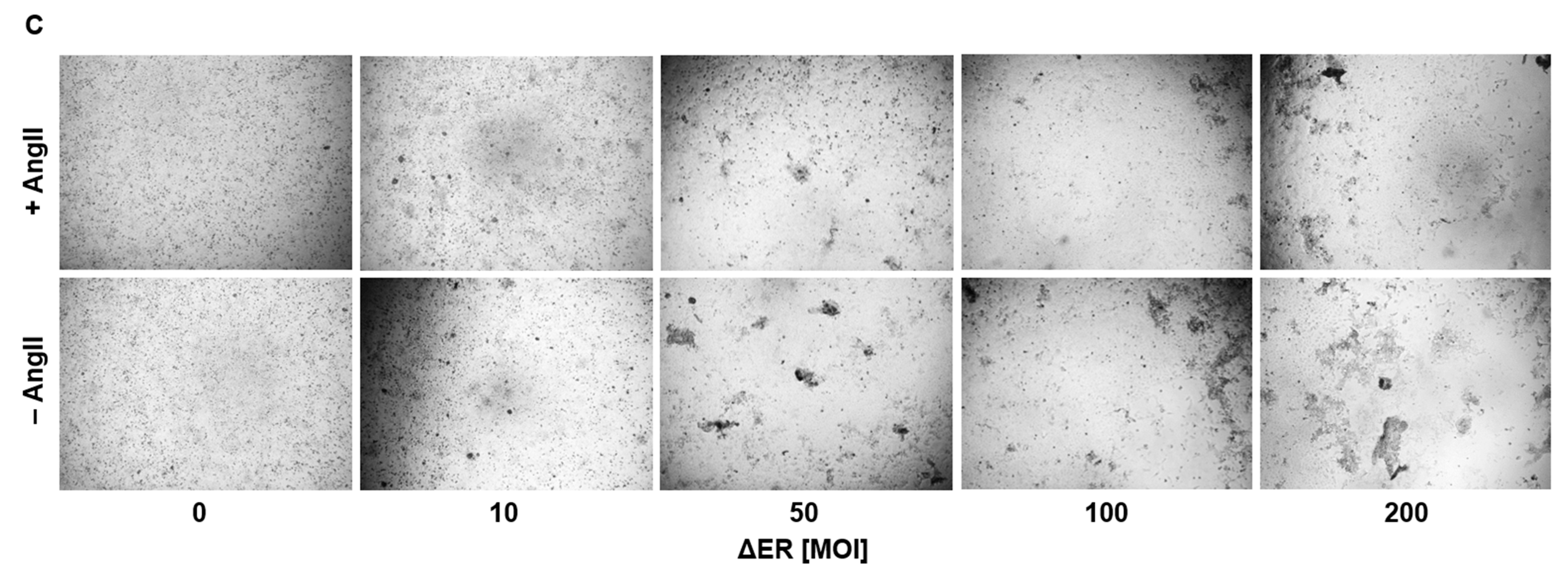
Prior to microscopic imaging, 3 × 104 ACE2-overexpressing U2OS cells were transduced with spike-expressing HAdV-C5 vectors with an MOI of 200 for 72 h. When analyzing the effect of angiotensin II (AngII) on syncytia formation, ACE2-overexpressing U2OS cells (3 × 104/well) were transduced with an MOI of 30–600 in the presence or absence of angiotensin II (2000 ng/well) for 72 h. Every 24 h, the medium was refreshed to consistently provide cells with AngII. Finally, 96-well plates were imaged using a Leica DMi8 S microscope.
2.15. Statistical Analysis
For statistical data analysis, the software RStudio was used. Normality was tested via the Shapiro–Wilk test and homogeneity of variance was investigated using Levene’s test. In the case of unequal variances, a Welch ANOVA was performed, and Dunnett’s T3 test was used as the post hoc test.
4. Discussion
With the conditional market approval for the emergency use of several adenoviral vector vaccines (e.g., Vaxzevria, Jcovden) during the COVID-19 pandemic, worldwide awareness of viral vector therapeutics rose. Adenoviral vectors proved themselves as a reliable platform for emergency outbreak diseases, being able to be quickly modified as well as manufactured at high rates while eliciting robust immune responses. While protein subunit vaccines and inactivated viral vaccines potentially require adjuvants for efficient stimulation of immune response, immunogenicity of adenoviral vectors achieves sufficient activation of the immune system on its own [
2,
12,
13]. Furthermore, while mRNA vaccines do not face splicing difficulties, their low stability demands storage at low temperatures, thus proving problematic for vaccination in developing countries.
On the contrary, certain adenoviral vector vaccines do not require cold chain storage [
2,
12,
13]. Overall, the adenoviral vector platform and the LNP-RNA platform do in fact complement each other rather than compete with each other. Still, knowledge regarding efficient adenoviral vaccine design is limited. Most of the market-approved vectors employ tetracycline-regulated protein expression for antigen production, masking possible transgene-related effects on patients, as these effects cannot be observed during propagation. Additionally, to date, there has been little experience pertaining to the incorporation of genes from an RNA virus into a DNA. As such, we strove to share the lessons we learned while generating our own spike protein-encoding vaccines based on human adenovirus type 5. In our study, we focused on investigating the effects of transgene codon optimization, expression cassette building blocks, and deletion or replacement of transgene internal functionalities on viral propagation and protein expression.
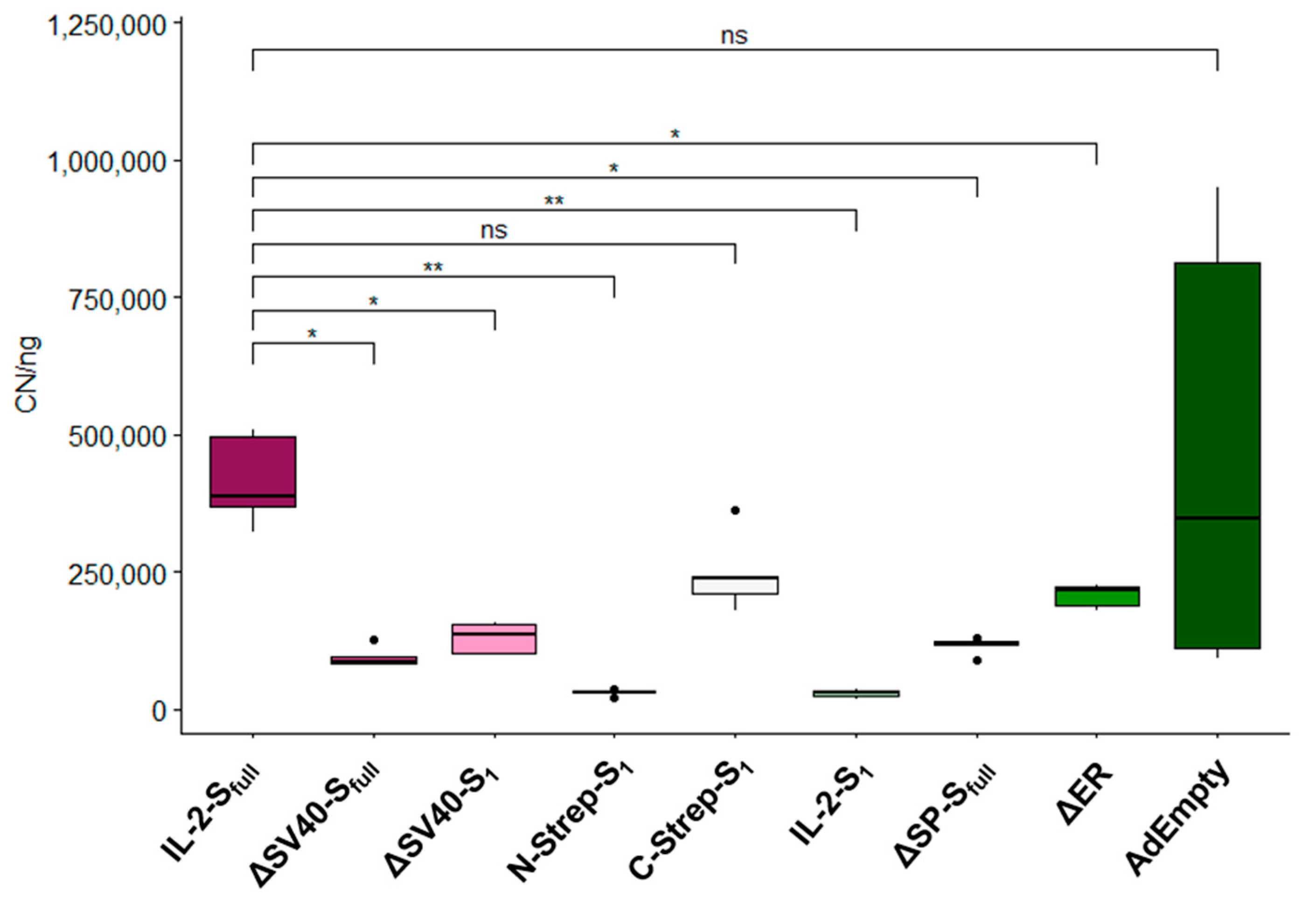
As our study shows, the design of such adenoviral vector vaccines for expressing a specific transgene is no trivial matter. A delicate balance between expression cassette functionality, viral propagation and protein expression level is necessary. We demonstrated the effect of expression cassette design on viral propagation, protein expression and mRNA splicing. As such, the use of SV40 intron in expression cassettes to enhance protein expression [
7] instead hampered the production of infectious viral particles considerably. Furthermore, the deletion of certain domains of the spike protein can influence infectivity and viral particle production, as observed for the ΔER vector. While deletion or replacement of the N-terminally located signal peptide of the spike protein did not seemingly interfere directly with viral propagation in this study, a precise statement cannot be provided, as vectors with deleted or replaced signal peptide also contained the SV40 intron. Still, our data suggest that careful consideration should be given to the insertion of additional sequences, such as introns or affinity tags, as minor adjustments may result in less infectious viral particles. Through qPCR analysis, these observations concerning viral propagation could be confirmed. Nearly all evaluated vectors showed a significant decrease in detected viral genomes compared to the IL-2-S
full vector.
Although vector growth did not appear to be affected for ∆SV40-S
full or ∆SV40-S
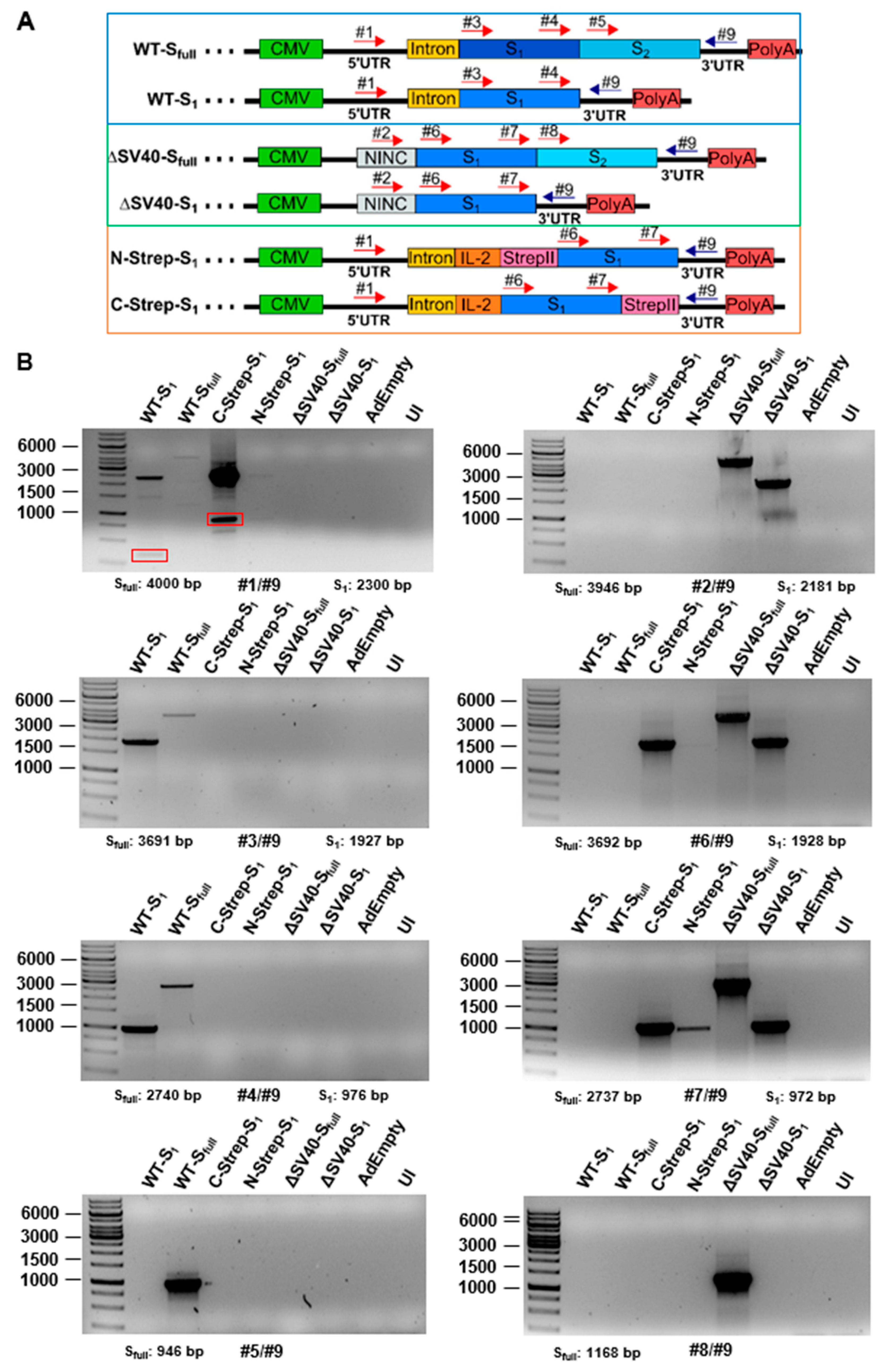
1 based on physical titers, viral particle production still seemed to be hampered according to qPCR results. As such, the expression of the spike protein may affect vector growth. As for protein expression analysis by Western blot, the spike protein was detected only for vectors expressing the codon-optimized version of the spike protein.Codon optimization of transgenes for vaccination approaches thus appears beneficial regarding protein expression level. Problems with mRNA splicing were expected to be the cause for undetectable spike protein expression because of SV40 intron inclusion in the ∆E1-located expression cassette. While additional bands could be identified when HEK293 cells were infected with vectors for one primer combination (#1/#9), no additional bands were observed when A549 cells were transduced. However, for most SV40 intron-containing vectors (such as WT-S
1, WT-S
full as well as C-Strep-S
1 vector), splice variants seem to exist. Sequencing of these additional bands (
Figure S2) suggests not only excision of introns from the processed mRNA but also the deletion of the IL-2 signal peptide and a major part of the S
1 protein. When predicting splice acceptor sites with the SpliceRover online tool [
14], a splice site (1473–1484 bp) could be predicted at the incised location. Similar results were obtained for the band at around 500 bp for the WT-S
1 vector construct (data not shown). As such, using expression-enhancing building blocks such as introns should be considered carefully, especially when transgenes contain splice acceptor sites. Transfer of RNA proteins into a DNA background or codon optimization may introduce additional splice sites, which should be considered for vaccine development. For codon optimization, it was previously determined that potential splice sites increased compared to the Wuhan isolate in ChAdOx-1 (Vaxzevria) and Ad26.COV2.S (Jcovden) in silico [
15]. The use of other introns, such as the HAdV-C5 tripartite leader intron instead of the SV40 intron, may alleviate the aberrant splicing we observed.
Furthermore, protein expression could not be detected for the secretion-modified IL-2-S
full vector. This could explain the growth behavior (
Figure 3) of this vector in comparison to the other vectors of this group (IL-2-S
1, ∆SP-S
full and ∆SP-S
1), which showed considerably hampered viral production (IL-2-S
1, ∆SP-S
full) or no viral particle production (∆SP-S
1). In general, vectors expressing increased levels of spike proteins, i.e., most of the vectors modified for their secretion profile (IL-2-S
1, ∆SP-S
full, ∆SP-S
1), appeared to be hampered in viral particle production as measured with physical titers (
Figure 2) and qPCR analysis (
Figure 3). As such, high expression of the spike protein also seems to affect viral propagation. This emphasizes the importance of balancing transgene expression and efficient viral vector production. Alternatively, transgene expression can be adjusted by choice of promoter [
16], a system to repress transgene expression during vector production [
17] or microRNA downregulation [
18,
19].
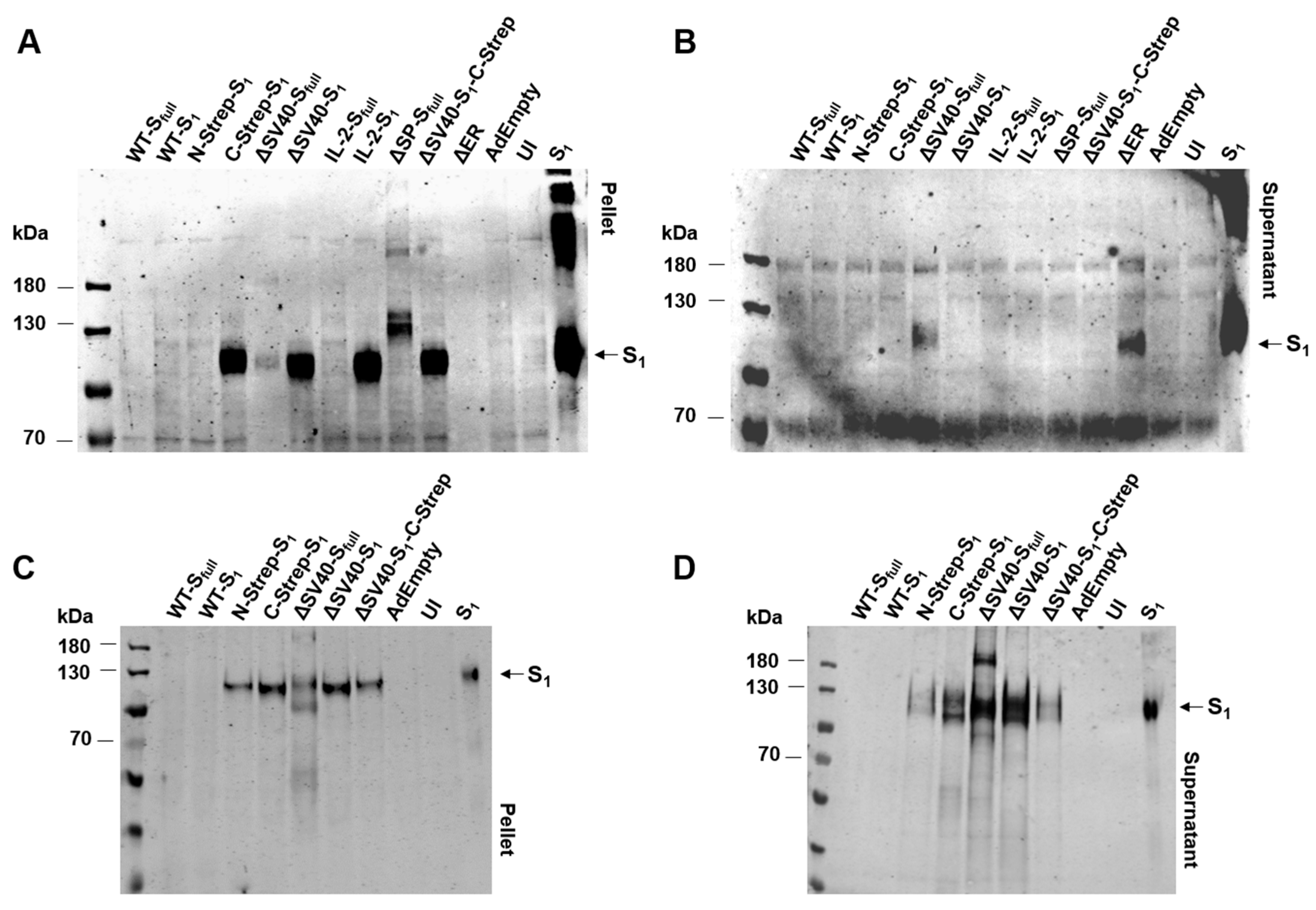
Unexpectedly, spike protein expression could not be detected for N-Strep-S
1 on Western blot analysis with anti-S
1 antibody (
Figure 4A,B). Only a very faint signal was detected when incubating with human serum after a prime boost with an mRNA vaccine. Contrary to this observation, a signal for C-Strep-S
1 and ΔSV40-S
1-C-Strep vectors could be determined by Western blot analysis (
Figure 4). As such, placement of Strep-tag at either the N- or C-terminus may also influence transgene protein expression. Previous studies clarified that placement of affinity tags at the N- or C-terminus should be considered carefully with regard to protein functionality and localization [
20]. As most signal peptides, including the signal peptide of the spike protein, are located at the N-terminus, placement of an affinity tag at C-terminus for spike protein expression to ensure proper localization seems appropriate.
Additionally, in this study, deletion of the ER retention signal seemed to promote syncytia formation in ACE2-overexpressing cell lines. A previous study also documented this increased formation of syncytia formation by ER retention signal-deleted vectors, when hACE2-mCherry cells were cocultured with S-∆19-EGFP-A549 (ER retention signal-deleted vector) [
21]. Compared to AdEmpty and ∆SV40-S
full, cell fusion between ACE2-expressing cells was increased when cells were transduced with the ∆ER vector.
This effect could also be observed in a panel of different cell lines, such as Huh-7, KM-12 and SH-SY5Y. Treatment with AngII compared to untreated control suggests involvement of ACE2 in the fusion process, as syncytia formation is decreased in AngII-treated cells. However, due to only performing a single experiment, a specific conclusion cannot be drawn, as further experiments would have to be performed. For example, ACE2 expression could be downregulated with miRNA and be compared with a nonsense miRNA control to observe whether ΔER vector-mediated syncytia formation would decrease.
To summarize, this study proved that the design of expression cassettes in adenoviral vaccine vectors can affect viral propagation and transgene protein expression. We observed that codon optimization of transgene sequence enhances protein expression, whereas deletion and/or replacement of protein domains can promote negative effects (e.g., fusogenicity) and negatively affect vector production. Expression-enhancing elements such as introns can prove harmful to viral infectivity. Splice sites may be generated by contextualizing an RNA gene into a DNA background or codon optimization. Therefore, for future adenoviral vaccine vector design, we emphasize the consideration of four main areas: (I) codon optimization, (II) careful consideration of deletion or replacement of transgene domains, (III) careful analyses of potential novel splice sites, and (IV) careful consideration of promoter choice and inclusion of, e.g., expression-enhancing elements such as introns. Nevertheless, one may still expect the unexpected.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







