
Accelerated Long-Fragment Circular PCR for Genetic Manipulation of Plant Viruses in Unveiling Functional Genomics
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construct, Target Gene, and Test Plant
2.2. Designing of Primers for Gene Insertion, Deletion, and Mutation
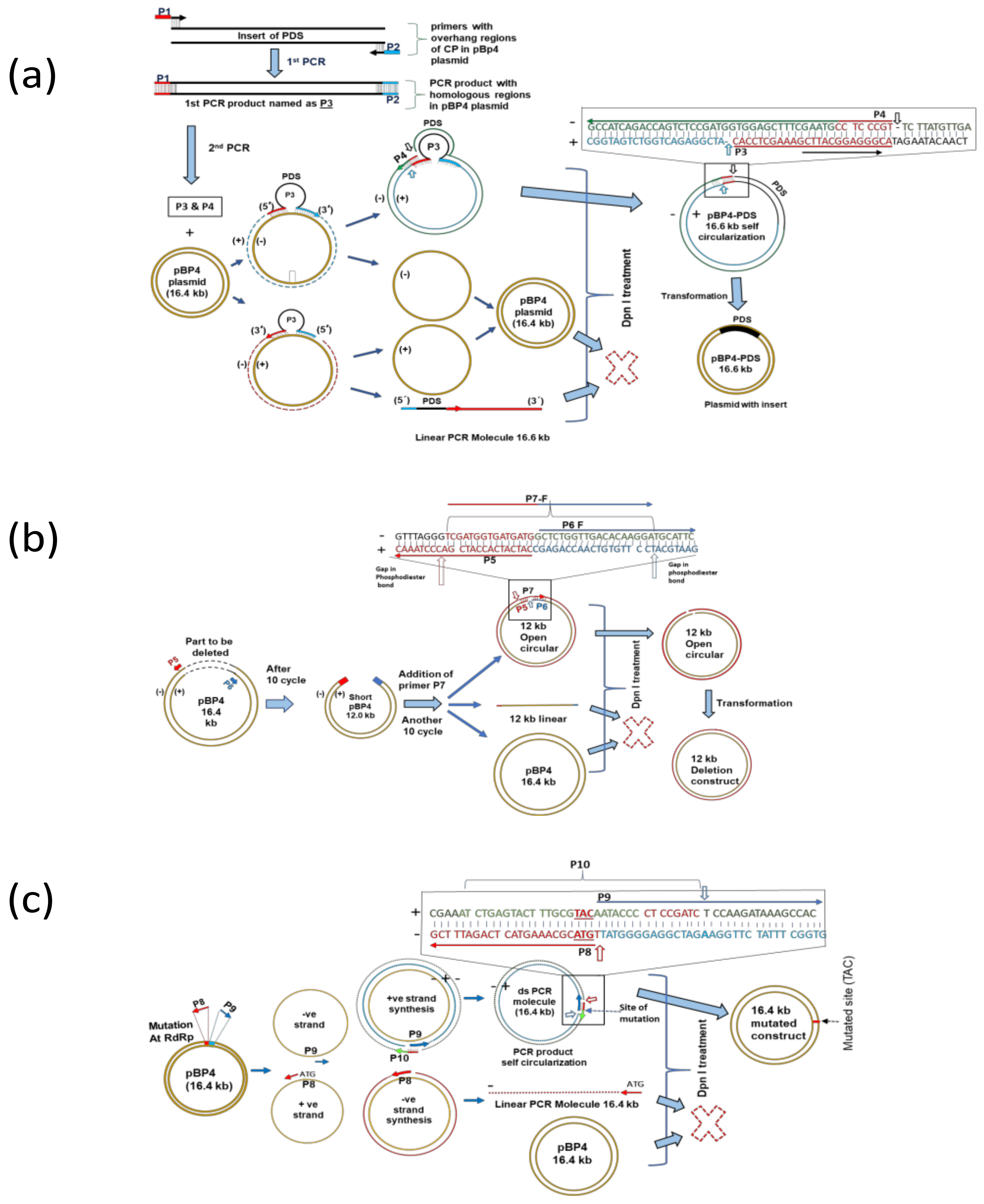
2.3. LC-PCR Protocol for Gene Insertion
2.4. LC-PCR Protocol for Deletion
2.5. LC-PCR Protocol for Substitution-Based Mutation
2.6. Agro Infiltration of the Constructs in Plant
2.7. In Planta Functional Assay
2.7.1. Detection of Virus Infectivity in the Plant by CGMMV Genome-Based Constructs
2.7.2. Determination of the Host Gene-Silencing and Associated Phenotypic Changes
2.7.3. Analysis of the in Planta Heterologous Protein Expression (Coat Protein of ToLCNDV)
2.7.4. Evaluation of the Role of Helicase and RNA Dependent RNA Polymerase Domain of CGMMV in Genome Replication
2.7.5. Assessment of the Infectivity of the CGMMV Mutant
3. Results
3.1. Construction of CGMMV Genome-Based VIGS Vector and Its Validation through Silencing of NbPDS Gene
3.2. Heterologous Expression of CPToLCNDV Gene Using CGMMV-Based Protein Expression Vector
3.3. Deletion of Helicase and RNA Dependent RNA Polymerase Restricts the Self-Replication of Deconstructed CGMMV Genome
3.4. Site-Directed Mutagenesis Leads to the Amino Acid Substitution in the CGMMV Genome and Subsequent the Abolition of Virus Symptoms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoenfeld, T.; Liles, M.; Wommack, K.E.; Polson, S.W.; Godisk, R.; Mead, D. Functional viral metagenomics and the next generation of molecular tools. Trends Microbiol. 2010, 18, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Han, S.S. Helical plant viral nanoparticles-bioinspired synthesis of nanomaterials and nanostructures. Bioinspir. Biomim. 2017, 12, 031001. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Han, S.S. Icosahedral plant viral nanoparticles—Bioinspired synthesis of nanomaterials/nanostructures. Adv. Colloid Interface Sci. 2017, 248, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.P.; Bonny, A.R.; Kumar, G.R.; Ng, A.H.; Town, J.; Wu, Q.C.; Aslankoohi, E.; Chen, S.Y.; Dods, G.; Harrigan, P.; et al. A Toolkit for rapid modular construction of biological circuits in mammalian cells. ACS Synth. Biol. 2019, 8, 2593–2606. [Google Scholar] [CrossRef]
- Caygill, R.L.; Blair, G.E.; Millner, P.A. A review on viral biosensors to detect human pathogens. Anal. Chim. Acta 2010, 681, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Bamogo, P.K.A.; Brugidou, C.; Sérémé, D.; Tiendrébéogo, F.; Djigma, F.W.; Simpore, J.; Lacombe, S. Virus-based pharmaceutical production in plants: An opportunity to reduce health problems in Africa. Virol. J. 2019, 16, 167. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Kwon, S.J.; Kim, M.H.; Choe, S.; Kwak, H.R.; Kim, M.K.; Jung, C.; Seo, J.K. A Plant Virus-Based Vector System for Gene Function Studies in Pepper. Plant Physiol. 2019, 181, 867–880. [Google Scholar] [CrossRef]
- Pasin, F.; Menzel, W.; Daròs, J.A. Harnessed viruses in the age of metagenomics and synthetic biology: An update on infectious clone assembly and biotechnologies of plant viruses. Plant Biotechnol. J. 2019, 17, 1010–1026. [Google Scholar] [CrossRef]
- Yang, S.J.; Revers, F.; Souche, S.; Lot, H.; Le Gall, O.; Candresse, T.; Dunez, J. Construction of full-length cDNA clones of lettuce mosaic virus (LMV) and the effects of intron-insertion on their viability in Escherichia coli and on their infectivity to plants. Arch. Virol. 1998, 143, 2443–2451. [Google Scholar] [CrossRef]
- López-Moya, J.J.; García, J.A. Construction of a stable and highly infectious introncontaining cDNA clone of plum pox potyvirus and its use to infect plants by particle bombardment. Virus Res. 2000, 68, 99–107. [Google Scholar] [CrossRef]
- Yamshchikov, V.; Mishin, V.; Cominelli, F. A new strategy in design of +RNA virus infectious clones enabling their stable propagation in E. coli. Virology 2001, 281, 272–280. [Google Scholar] [CrossRef]
- Ali, M.C.; Omar, A.S.; Natsuaki, T. An infectious full-length cDNA clone of potato virus YNTN-NW, a recently reported strain of PVY that causes potato tuber necrotic ringspot disease. Arch. Virol. 2011, 156, 2039–2043. [Google Scholar] [CrossRef]
- Li, D.; Aaskov, J.; Lott, W.B. Identification of a cryptic prokaryotic promoter within the cDNA encoding the 5′ end of Dengue virus RNA genome. PLoS ONE 2011, 6, e18197. [Google Scholar] [CrossRef]
- DeKeyser, J.M.; Thompson, C.H.; George, A.L., Jr. Cryptic prokaryotic promoters explain instability of recombinant neuronal sodium channels in bacteria. J. Biol. Chem. 2021, 296, 100298. [Google Scholar] [CrossRef]
- Tillett, D.; Neilan, B.A. Enzyme-free cloning: A rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999, 27, e26. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Throop, A.L.; LaBaer, J. Site-specific recombinational cloning using Gateway and In-Fusion cloning schemes. Curr. Protoc. Mol. Biol. 2015, 110, 3–20. [Google Scholar] [CrossRef]
- Lu, Q. Seamless cloning and gene fusion. Trends Biotechnol. 2005, 23, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chee, J.Y.; Chin, C.F. Gateway cloning technology: Advantages and drawbacks. Cloning Transgenes 2015, 4, 138. [Google Scholar] [CrossRef]
- Quan, J.; Tian, J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS ONE 2009, 4, e6441. [Google Scholar] [CrossRef]
- Quan, J.; Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011, 6, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Liu, C.J.; Wu, L.; Wu, J.F.; Yin, X.N.; Deng, K.H.; Zhang, D.Y.; Meng, E. MCT cloning: A seamless cloning strategy for inserting DNA fragments. Biotechnol. Biotechnol. Equip. 2018, 32, 1298–1305. [Google Scholar] [CrossRef]
- Spiliotis, M. Inverse Fusion PCR Cloning. PLoS ONE 2012, 7, e35407. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, A.; Andersen, K.R.; Schwartz, T.U. Exponential megapriming PCR (EMP) cloning-seamless DNA insertion into any target plasmid without sequence constraints. PLoS ONE 2012, 7, e53360. [Google Scholar] [CrossRef]
- Liu, C.J.; Jiang, H.; Wu, L.; Zhu, L.Y.; Meng, E.; Zhang, D.Y. OEPR cloning: An efficient and seamless cloning strategy for large-and multi-fragments. Sci. Rep. 2017, 7, 44648. [Google Scholar] [CrossRef]
- Zeng, F.; Zang, J.; Zhang, S.; Hao, Z.; Dong, J.; Lin, Y. AFEAP cloning: A precise and efficient method for large DNA sequence assembly. BMC Biotechnol. 2017, 17, 81. [Google Scholar] [CrossRef]
- Guo, W.; Xie, B.; Jiang, M.; Zhu, X.J.; Qiu, M.; Dai, Z.M. An improved overlap extension PCR for simultaneous multiple sites large fragments insertion, deletion and substitution. Sci. Rep. 2019, 9, 15637. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef]
- Zeng, F.; Zhang, S.; Hao, Z.; Duan, S.; Meng, Y.; Li, P.; Dong, J.; Lin, Y. Efficient strategy for introducing large and multiple changes in plasmid DNA. Sci. Rep. 2018, 8, 1714. [Google Scholar] [CrossRef]
- Liu, M.; Liang, Z.; Aranda, M.A.; Hong, N.; Liu, L.; Kang, B.; Gu, Q. A cucumber green mottle mosaic virus vector for virus-induced gene silencing in cucurbit plants. Plant Methods 2020, 16, 9. [Google Scholar] [CrossRef]
- Jailani, A.A.K.; Solanki, V.; Roy, A.; Sivasudha, T.; Mandal, B. A CGMMV genome-replicon vector with partial sequences of coat protein gene efficiently expresses GFP in Nicotiana benthamiana. Virus Res. 2017, 233, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.H.; Chen, B.; Chen, H.; Menassa, R.; Hao, X.; Bernards, M.; Hüner, N.P.A.; Wang, A. Development of a cucumber green mottle mosaic virus-based expression vector for the production in cucumber of neutralizing epitopes against a devastating animal virus. J. Virol. Methods 2019, 269, 18–25. [Google Scholar] [CrossRef]
- Jailani, A.A.; Hendricks, K.; Roberts, P.D.; Paret, M.L. Development of a simple one-step multiplex RT-PCR system for simultaneous detection of DNA and RNA viruses of cucurbit leaf crumple virus, cucurbit yellow stunting disorder virus, squash vein yellowing virus, and cucurbit chlorotic yellows virus. Physiol. Mol. Plant Pathol. 2021, 116, 101734. [Google Scholar] [CrossRef]
- Jailani, A.A.; Iriarte, F.; Hochmuth, B.; Willis, S.M.; Warren, M.; Dey, K.; Velez-Climent, M.; McVay, J.; Bag, S.; Paret, M.L. First report of cucurbit chlorotic yellows virus affecting watermelon in the United States. Plant Dis. 2022, 106, 774. [Google Scholar] [CrossRef] [PubMed]
- Jailani, A.A.K.; Iriarte, F.; Paret, M.L. First report of watermelon crinkle leaf-associated virus (WCLaV)-1 and WCLaV-2 infecting straightneck squash in the United States. Plant Dis. 2023, 107, 2561. [Google Scholar] [CrossRef] [PubMed]
- Jailani, A.A.K.; Paret, M.L. Development of a Multiplex RT-RPA Assay for Simultaneous Detection of Three Viruses in Cucurbits. Mol. Plant Pathol. 2023, 24, 1443–1450. [Google Scholar] [CrossRef]
- Weigel, D.; Glazebrook, J. Transformation of agrobacterium using the freeze-thaw method. CSH Protoc. 2006, 2006, pdb-prot4666. [Google Scholar] [CrossRef]
- Leuzinger, K.; Dent, M.; Hurtado, J.; Stahnke, J.; Lai, H.; Zhou, X.; Chen, Q. Efficient agroinfiltration of plants for high-level transient expression of recombinant proteins. J. Vis. Exp. 2013, 77, 50521. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Jailani, A.A.K.; Shilpi, S.; Mandal, B. Rapid demonstration of infectivity of a hybrid strain of potato virus Y occurring in India through overlapping extension PCR. Physiol. Mol. Plant Pathol. 2017, 98, 62–68. [Google Scholar] [CrossRef]
- Jailani, A.A.K.; Kumar, A.; Mandal, B.; Sivasudha, T.; Roy, A. Agroinfection of tobacco by croton yellow vein mosaic virus and designing of a replicon vector for expression of foreign gene in plant. Virusdisease 2016, 27, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Bryksin, A.V.; Matsumura, I. Overlap extension PCR cloning: A simple and reliable way to create recombinant plasmids. Biotechniques 2010, 48, 463–465. [Google Scholar] [CrossRef]
- Lee, L.Y.; Gelvin, S.B. T-DNA binary vectors and systems. Plant Physiol. 2008, 146, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Songkumarn, P.; Liu, J.; Wang, G.L. A versatile zero background T-vector system for gene cloning and functional genomics. Plant Physiol. 2009, 150, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Wei, X.M.; Ye, X.D.; Xu, H.X.; Zhou, X.P.; Liu, S.S.; Wang, X.W. Expression and functional characterisation of a soluble form of Tomato yellow leaf curl virus coat protein. Pest Manag. Sci. 2014, 70, 1624–1631. [Google Scholar] [CrossRef]
- Elgaied, L.; Salem, R.; Elmenofy, W. Expression of tomato yellow leaf curl virus coat protein using baculovirus expression system and evaluation of its utility as a viral antigen. 3 Biotech 2017, 7, 269. [Google Scholar] [CrossRef]
- Lorenz, T.C. Polymerase chain reaction: Basic protocol plus troubleshooting and optimization strategies. J. Vis. Exp. 2012, 63, e3998. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequences | Used for | Purpose |
|---|---|---|---|
| P1-F | CACCTCGAAAGCTTAGGGAGGGCAATCTTATGTTGA | Amplification of NbPDS gene from N. benthamiana | Development of pBP4-NbPDS constructs for silencing of NbPDS gene |
| P2-R | ATCAGAAGACCCTCGAAAGGAGGGTTACCATCTAAAA | ||
| P3 | 1st PCR product as mega primer | Synthesis of pBP4 plasmid with the NbPDS sequence | |
| P4 | TGCCCTCCCTAAGCTTTCGAGGTGGTAGCCTCTGACCAGACTACCG | Self-circularisation of pBP4 with NbPDS sequence | |
| P1a-F | GCTTCACAAGGTACCGCTATGGCGAAGCGACCAGCAGAT | Amplification of capsid protein (CP) gene of ToLCNDV | Development of pBP4-∆CP-CPToLCNDV for the expression of ToLCNDV CP gene |
| P2b-R | ATTTGTGACCGAATCATAAAAATAGCACCATCAGAAGACCCTCGAAACTA | ||
| P3c | 1st PCR product as mega primer | Synthesis of pBP4-∆CP with the CP gene of ToLCNDV | |
| P4d | CGCTTCGCCATAGCGGTACCTTGTGAAGCAACTAGAAAATTAAG | Self-circularisation of pBP4-∆CP with the CP gene of ToLCNDV | |
| P5-R | CATCATCACCATCGACCCTAAACTG | Synthesis of full-length pBP4 from the deletion ends | Development of pBP4-∆Rep for the deletion of helicase and RNA-dependent RNA Polymerase gene from CGMMV genome |
| P6-F | GCTCTGGTTGACACAAGGATGCATTC | ||
| P7-F | TCGATGGTGATGATGGCTCTGGTTGACACAAGGA | Self-circularisation of newly synthesized strands in pBP4 construct excluding the deleted Repportion | |
| P8-F | GTACGCAAAGTACTCAGATTTCGATTTC | Amplification of full-length BP4 with a terminal mutation at 1066th-68th nucleotide position | Creation of mutant CGMMV construct (pBP4-∆1066-68) |
| P9-R | AATACCCCTCCGATCTTCCAAGATAAAGCCAC | ||
| P10-R | CGAAATCTGAGTACTTTGCGTACAATACCCCTCCGATC | Self-circularisation of pBP4 with mutation from CTG (leucine) to TAC (tyrosin) |
| Manipulation Strategy | Purpose | Size Manipulated (nt) | Total Size of Construct (Kb) | No. of Attempts | Average No. of Colonies | No Positive Colonies/ Tested by PCR | Success (%) |
|---|---|---|---|---|---|---|---|
| Insertion of NbPDS gene | Gene silencing | 21 0 | 16.6 | 5 | 95 | 24/25 | 96 |
| Insertion of CPToLCNDVgene | Protein Expression | 770 | 16.6 | 10 | 80 | 22/25 | 88 |
| Deletion | Viral Replication | 4400 | 12.0 | 3 | 150 | 25/25 | 100 |
| Mutation | Phenotype | 1 | 16.4 | 5 | 200 | 25/25 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jailani, A.A.K.; Chattopadhyay, A.; Kumar, P.; Singh, O.W.; Mukherjee, S.K.; Roy, A.; Sanan-Mishra, N.; Mandal, B. Accelerated Long-Fragment Circular PCR for Genetic Manipulation of Plant Viruses in Unveiling Functional Genomics. Viruses 2023, 15, 2332. https://doi.org/10.3390/v15122332
Jailani AAK, Chattopadhyay A, Kumar P, Singh OW, Mukherjee SK, Roy A, Sanan-Mishra N, Mandal B. Accelerated Long-Fragment Circular PCR for Genetic Manipulation of Plant Viruses in Unveiling Functional Genomics. Viruses. 2023; 15(12):2332. https://doi.org/10.3390/v15122332
Chicago/Turabian StyleJailani, A. Abdul Kader, Anirudha Chattopadhyay, Pradeep Kumar, Oinam Washington Singh, Sunil Kumar Mukherjee, Anirban Roy, Neeti Sanan-Mishra, and Bikash Mandal. 2023. "Accelerated Long-Fragment Circular PCR for Genetic Manipulation of Plant Viruses in Unveiling Functional Genomics" Viruses 15, no. 12: 2332. https://doi.org/10.3390/v15122332
APA StyleJailani, A. A. K., Chattopadhyay, A., Kumar, P., Singh, O. W., Mukherjee, S. K., Roy, A., Sanan-Mishra, N., & Mandal, B. (2023). Accelerated Long-Fragment Circular PCR for Genetic Manipulation of Plant Viruses in Unveiling Functional Genomics. Viruses, 15(12), 2332. https://doi.org/10.3390/v15122332