
An Estuarine Cyanophage S-CREM1 Encodes Three Distinct Antitoxin Genes and a Large Number of Non-Coding RNA Genes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Host Incubation and Cyanophage Isolation
2.2. Host Range Determination
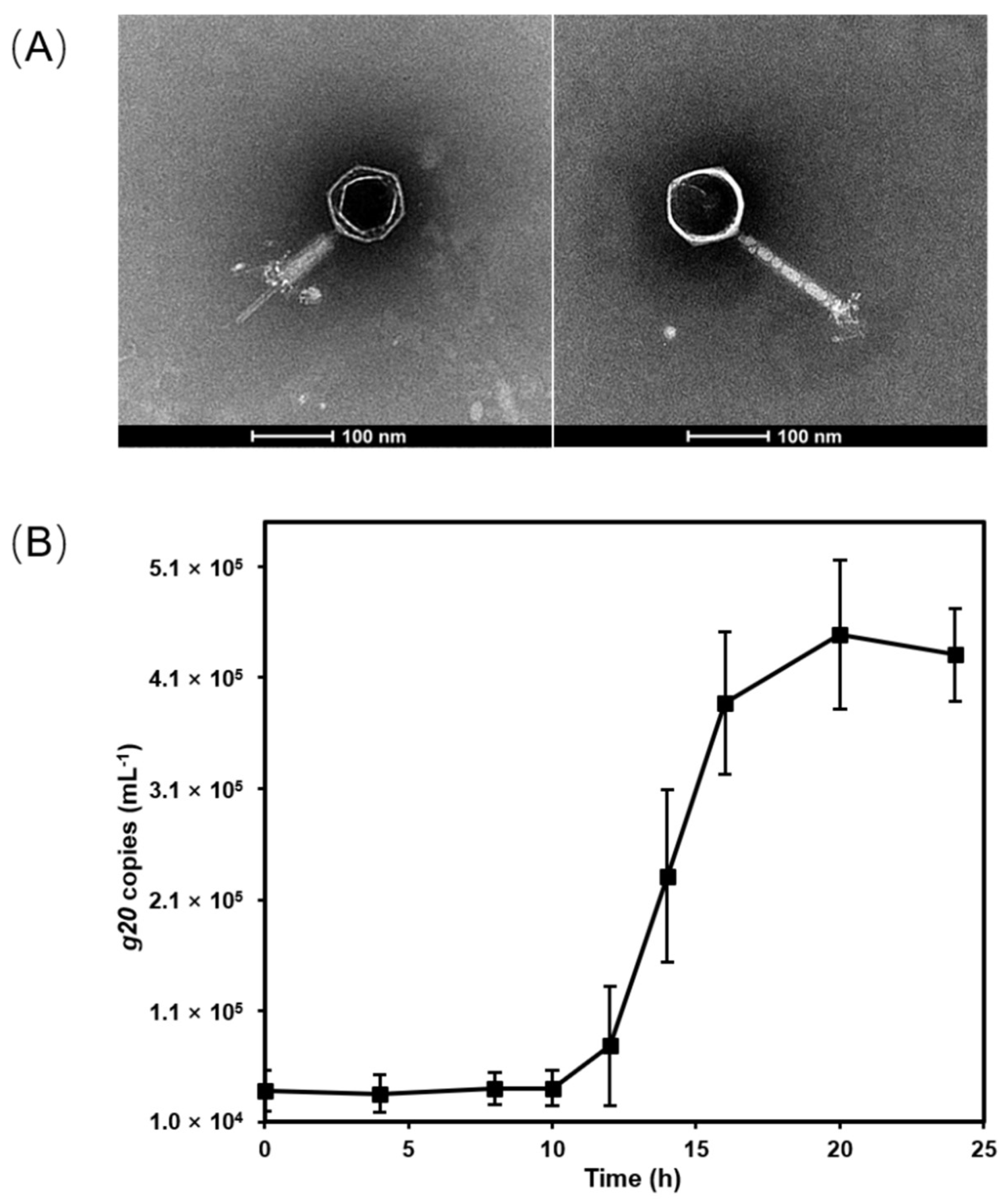
2.3. One-Step Growth Curve
2.4. Phage Amplification and Purification
2.5. Transmission Electron Microscopy (TEM) Observation
2.6. Phage Genome DNA Extraction and Sequencing
2.7. Genomic and Phylogenetic Analyses
2.8. Identification of the S-CREM1 Virion Proteins by Mass Spectrometry
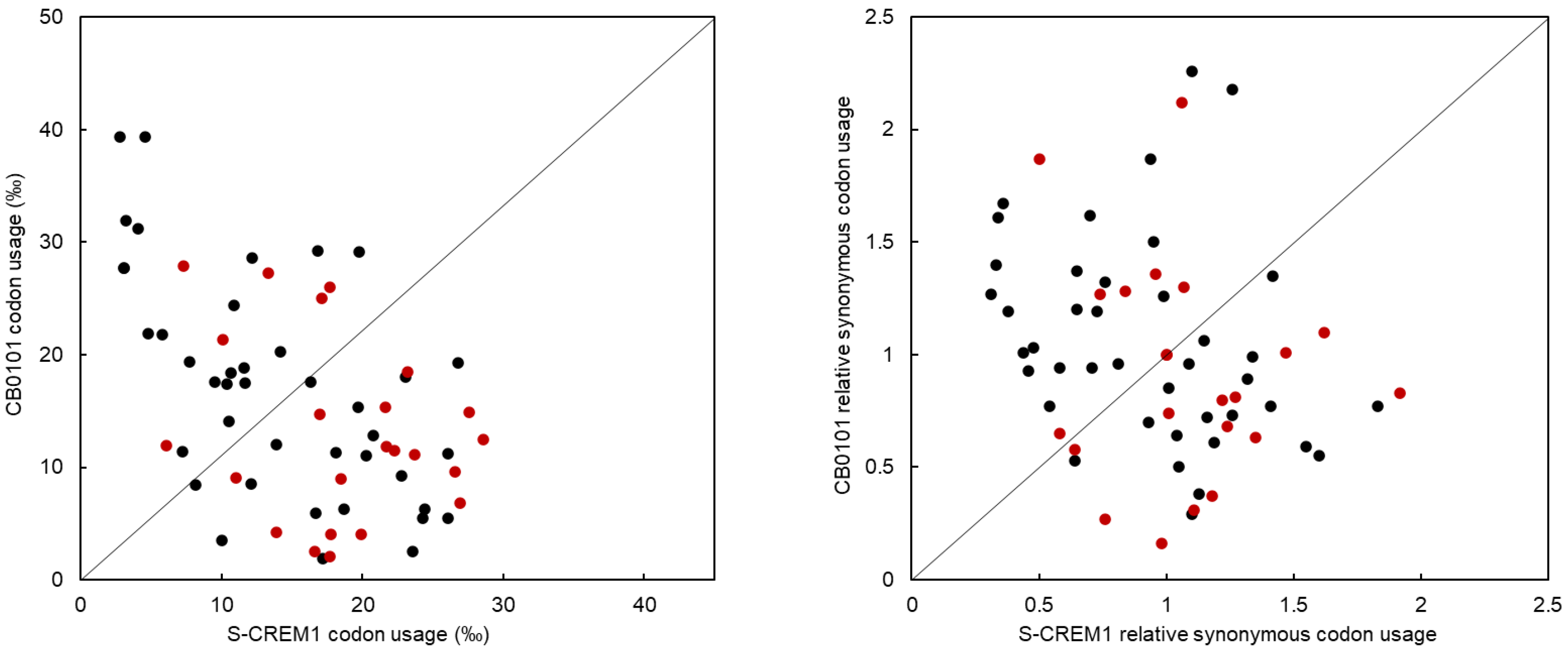
2.9. Codon Usage (CU) and Relative Synonymous Codon Usage (RSCU) Analyses
2.10. Motifs Prediction of sRNA and cis-Regulatory RNA Genes
3. Results and Discussion
3.1. General Features of S-CREM1
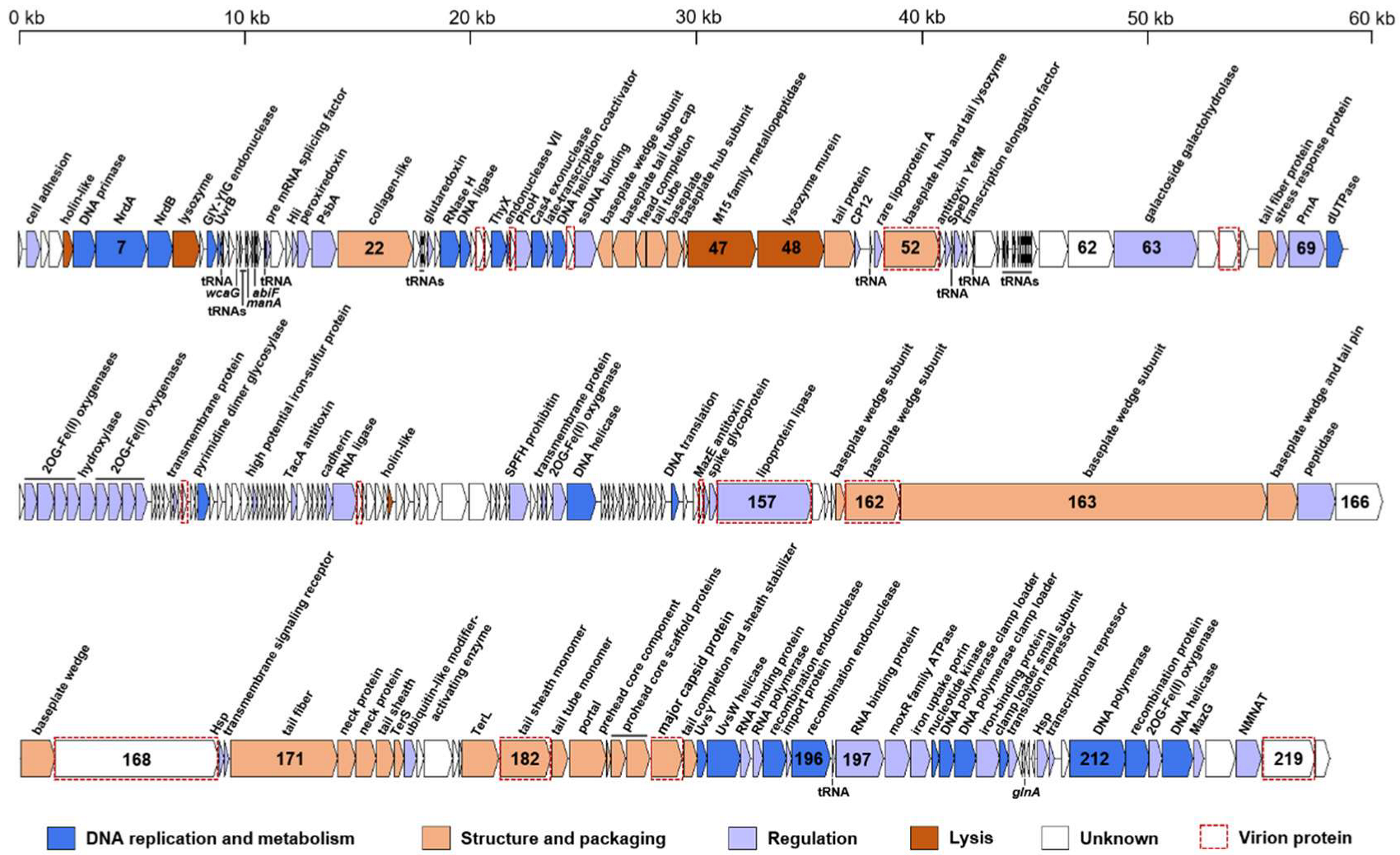
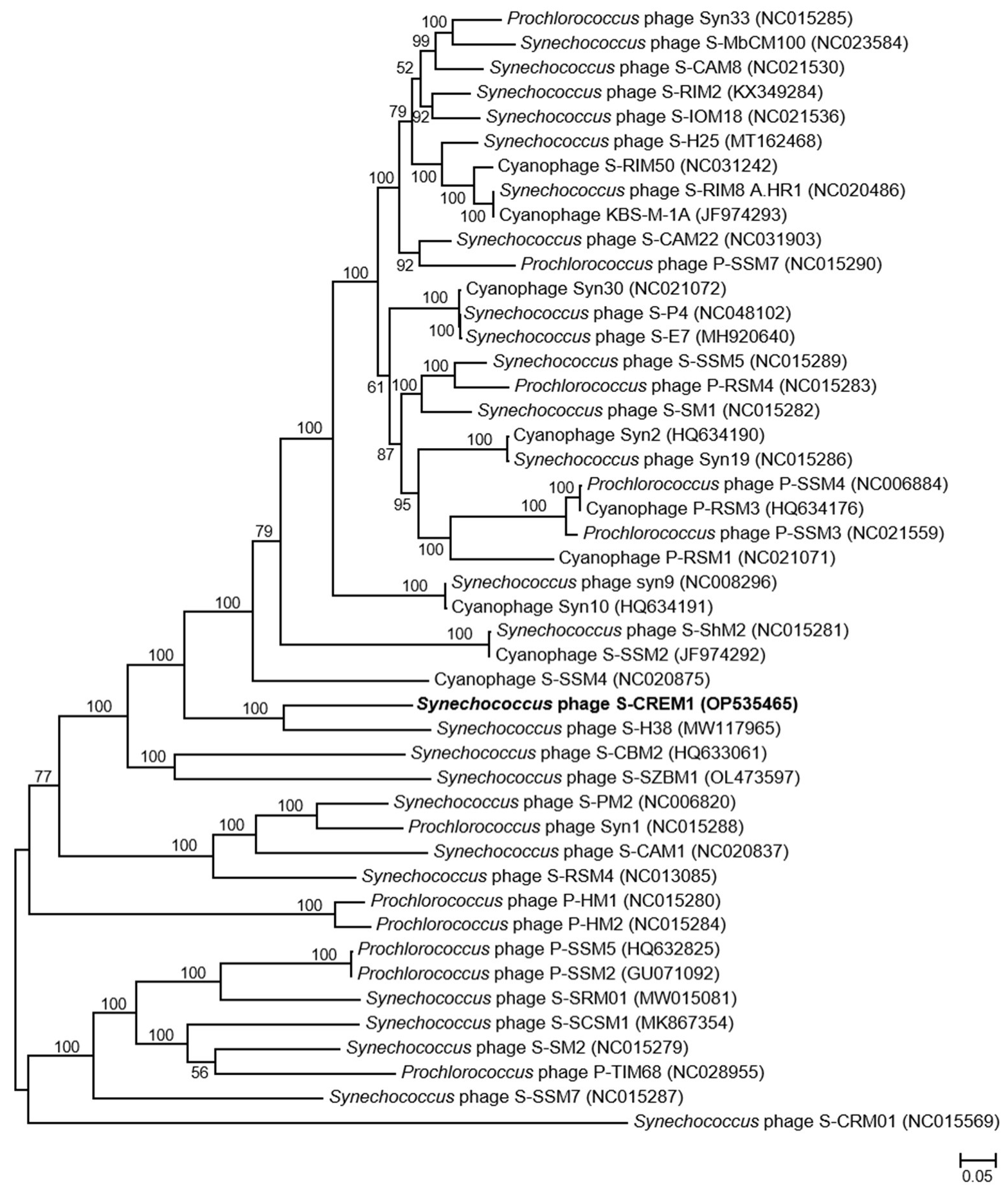
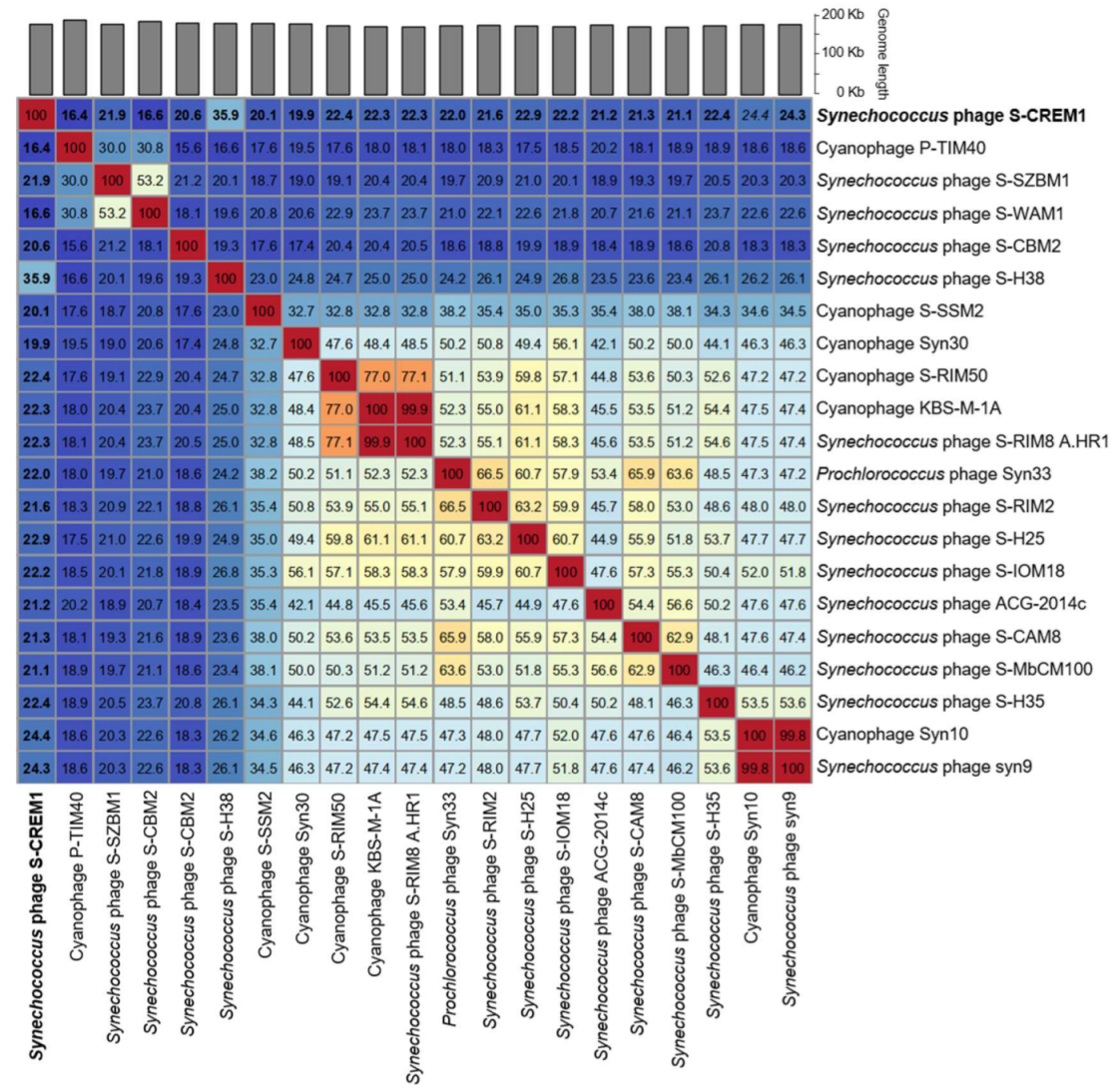
3.2. Genomic Features of S-CREM1 and Proposal of a New Viral Genus
3.3. Various and Unique AMGs in the S-CREM1 Genome
3.3.1. Cyanophage-Featured AMGs
3.3.2. Three Antitoxin Genes
3.3.3. A MoxR Family ATPase Gene
3.3.4. Overlooked Pyrimidine Dimer DNA Glycosylase Genes in Cyanophages
3.4. A Large Number of tRNA Genes
3.5. One Small RNA (sRNA) and Three cis-Regulatory RNA Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Partensky, F.B.J.; Vaulot, D. Differential distribution and ecology of Prochlorococcus and Synechococcus in oceanic waters: A review. Bull. Inst. Océanogr. 1999, 19, 457–475. [Google Scholar]
- Callieri, C.; Cronberg, G.; Stockner, J.G. Freshwater Picocyanobacteria: Single cells, Microcolonies and Colonial Forms. In Ecology of Cyanobacteria II; Whitton, B.A., Ed.; Springer: Amsterdam, The Netherlands, 2012; pp. 229–269. [Google Scholar]
- Affronti, L.F.; Marshall, H.G. Using frequency of dividing cells in estimating autotrophic picoplankton growth and productivity in the Chesapeake Bay. Hydrobiologia 1994, 284, 193–203. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef]
- Wang, K.; Chen, F. Prevalence of highly host-specific cyanophages in the estuarine environment. Environ. Microbiol. 2008, 10, 300–312. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Cai, L.; Zhang, R.; Wei, S.; Li, F.; Liu, Y.; Xu, Y. A unique set of auxiliary metabolic genes found in an isolated cyanophage sheds new light on marine phage–host interactions. Microbiol. Spectr. 2022, 10, e02367-22. [Google Scholar] [CrossRef]
- Lindell, D.; Sullivan, M.B.; Johnson, Z.I.; Tolonen, A.C.; Rohwer, F.; Chisholm, S.W. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc. Natl. Acad. Sci. USA 2004, 101, 11013–11018. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, B.; Xue, B.; Lundin, D.; Edwards, R.A.; Breitbart, M. A bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol. Biol. 2013, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Enav, H.; Mandel-Gutfreund, Y.; Béjà, O. Comparative metagenomic analyses reveal viral-induced shifts of host metabolism towards nucleotide biosynthesis. Microbiome 2014, 2, 9. [Google Scholar] [CrossRef]
- Kelly, L.; Ding, H.; Huang, K.H.; Osburne, M.S.; Chisholm, S.W. Genetic diversity in cultured and wild marine cyanomyoviruses reveals phosphorus stress as a strong selective agent. ISME J. 2013, 7, 1827–1841. [Google Scholar] [CrossRef] [Green Version]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef]
- Enav, H.; Béjà, O.; Mandel-Gutfreund, Y. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 2012, 6, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Fang, W.; Miranda-Sanchez, F.; Brown, J.M.; Kauffman, K.M.; Acevero, C.M.; Bartel, D.P.; Polz, M.F.; Kelly, L. Degradation of host translational machinery drives tRNA acquisition in viruses. Cell Syst. 2021, 12, 771–779.e5. [Google Scholar] [CrossRef]
- Weinberg, Z.; Wang, J.X.; Bogue, J.; Yang, J.; Corbino, K.; Moy, R.H.; Breaker, R.R. Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010, 11, R31. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [Green Version]
- Field, C.B.; Behrenfeld, M.J.; Randerson, J.T.; Falkowski, P. Primary Production of the Biosphere: Integrating Terrestrial and Oceanic Components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhan, Y.; Marsan, D.; Páez-Espino, D.; Cai, L.; Chen, F. Uncultivated viral populations dominate estuarine viromes on the spatiotemporal scale. mSystems 2021, 6, e01020-20. [Google Scholar] [CrossRef]
- Fucich, D.; Marsan, D.; Sosa, A.; Chen, F. Complete genome sequence of subcluster 5.2 Synechococcus sp. strain CB0101, isolated from the Chesapeake Bay. Microbiol. Resour. Announc. 2019, 8, e00484-19. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, R.; Wang, N.; Cai, L.; Tong, Y.; Sun, Q.; Chen, F.; Jiao, N. Novel phage–host interactions and evolution as revealed by a cyanomyovirus isolated from an estuarine environment. Environ. Microbiol. 2018, 20, 2974–2989. [Google Scholar] [CrossRef] [PubMed]
- Dreher, T.W.; Brown, N.; Bozarth, C.S.; Schwartz, A.D.; Riscoe, E.; Thrash, C.; Bennett, S.E.; Tzeng, S.C.; Maier. C.S. A freshwater cyanophage whose genome indicates close relationships to photosynthetic marine cyanomyophages. Environ. Microbiol. 2011, 13, 1858–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, N.H.; Clokie, M.R.J.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The Genome of S-PM2, a “Photosynthetic” T4-Type Bacteriophage That Infects Marine Synechococcus Strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Gao, C.; Jiang, T.; You, S.; Guo, C.; He, H.; Liu, Y.; Zhang, X.; Shao, H.; Liu, H.; et al. Genomic analysis of Synechococcus phage S-B43 and its adaption to the coastal environment. Virus Res. 2020, 289, 198155. [Google Scholar] [CrossRef]
- Jiang, T.; Guo, C.; Wang, M.; Wang, M.; Zhang, X.; Liu, Y.; Liang, Y.; Jiang, Y.; He, H.; Shao, H.; et al. Genome Analysis of Two Novel Synechococcus Phages that Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses. Viruses 2020, 12, 800. [Google Scholar] [CrossRef]
- Marston, M.F.; Martiny, J. Genomic diversification of marine cyanophages into stable ecotypes. Environ. Microbiol. 2016, 18, 4240–4253. [Google Scholar] [CrossRef]
- Stoddard, L.I.; Martiny, J.B.H.; Marston, M.F. Selection and Characterization of Cyanophage Resistance in Marine Synechococcus Strains. Appl. Environ. Microbiol. 2007, 73, 5516–5522. [Google Scholar] [CrossRef] [Green Version]
- Waterbury, J.; Watson, S.; Valois, F.; Franks, D. Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can. J. Fish. Aquat. Sci. 1986, 214, 71–120. [Google Scholar]
- Chen, F.; Wang, K.; Kan, J.; Bachoon, D.S.; Lu, J.; Lau, S.; Campbell, L. Phylogenetic diversity of Synechococcus in the Chesapeake Bay revealed by ribulose-1, 5-bisphosphate carboxylase-oxygenase (RuBisCO) large subunit gene (rbcL) sequences. Aquat. Microb. Ecol. Int. J. 2004, 36, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; Joint, I.R.; Carr, N.G.; Mann, N.H. Isolation and molecular characterization of five marine cyanophages propagated on Synechococcus sp. strain WH7803. Appl. Environ. Microbiol. 1993, 59, 3736–3743. [Google Scholar] [CrossRef] [Green Version]
- Ciglenečki, U.J.; Grom, J.; Toplak, I.; Jemeršić, L.; Barlič-Maganja, D. Real-time RT-PCR assay for rapid and specific detection of classical swine fever virus: Comparison of SYBR Green and TaqMan MGB detection methods using novel MGB probes. J. Virol. Methods 2008, 147, 257–264. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, B.; Xu, Y.; Zhang, Z.; Man, H.; Liu, J.; Chen, F. An Inducible Microbacterium Prophage vB_MoxS-R1 Represents a Novel Lineage of Siphovirus. Viruses 2022, 14, 731. [Google Scholar] [CrossRef]
- Ma, R.; Lai, J.; Chen, X.; Wang, L.; Yang, Y.; Wei, S.; Jiao, N.; Zhang, R. A Novel Phage Infecting Alteromonas Represents a Distinct Group of Siphophages Infecting Diverse Aquatic Copiotrophs. Msphere 2021, 6, e0045421. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Doron, S.; Fedida, A.; Hernández-Prieto, M.A.; Sabehi, G.; Karunker, I.; Stazic, D.; Feingersch, R.; Steglich, C.; Futschik, M.; Lindell, D.; et al. Transcriptome dynamics of a broad host-range cyanophage and its hosts. ISME J. 2015, 10, 1437–1455. [Google Scholar] [CrossRef] [Green Version]
- Rong, C.; Zhou, K.; Li, S.; Xiao, K.; Xu, Y.; Zhang, R.; Yang, Y.; Zhang, Y. Isolation and Characterization of a Novel Cyanophage Encoding Multiple Auxiliary Metabolic Genes. Viruses 2022, 14, 887. [Google Scholar] [CrossRef]
- Xu, Y.; Jiao, N.; Chen, F. Novel psychrotolerant picocyanobacteria isolated from Chesapeake Bay in the winter. J. Phycol. 2015, 51, 782–790. [Google Scholar] [CrossRef]
- Mackey, K.R.; Paytan, A.; Caldeira, K.; Grossman, A.R.; Moran, D.; McIlvin, M.; Saito, M.A. Effect of Temperature on Photosynthesis and Growth in Marine Synechococcus spp. Plant Physiol. 2013, 163, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Toledo, G.; Palenik, B. Synechococcus diversity in the California current as seen by RNA polymerase (rpoC1) gene sequences of isolated strains. Appl. Environ. Microbiol. 1997, 63, 4298–4303. [Google Scholar] [CrossRef] [Green Version]
- Palenik, B.; Brahamsha, B.; Larimer, F.W.; Land, M.; Hauser, L.; Chain, P.; Lamerdin, J.; Regala, W.; Allen, E.E.; McCarren, J.; et al. The genome of a motile marine Synechococcus. Nature 2003, 424, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Gao, E.-B.; Huang, Y.; Ning, D. Metabolic Genes within Cyanophage Genomes: Implications for Diversity and Evolution. Genes 2016, 7, 80. [Google Scholar] [CrossRef]
- Callaghan, M.M.; Amador-Noguez, D. Hostile Takeover: How Viruses Reprogram Prokaryotic Metabolism. Annu. Rev. Microbiol. 2021, 75, 515–539. [Google Scholar] [CrossRef]
- Karabencheva-Christova, T.G.; Torras, J.; Mulholland, A.J.; Lodola, A.; Christov, C.Z. Mechanistic Insights into the Reaction of Chlorination of Tryptophan Catalyzed by Tryptophan 7-Halogenase. Sci. Rep. 2017, 7, 17395. [Google Scholar] [CrossRef] [Green Version]
- van Staalduinen, L.M.; Jia, Z. Post-translational hydroxylation by 2OG/Fe(II)-dependent oxygenases as a novel regulatory mechanism in bacteria. Front. Microbiol. 2015, 5, 798. [Google Scholar] [CrossRef] [Green Version]
- LeRoux, M.; Laub, M.T. Toxin-Antitoxin Systems as Phage Defense Elements. Annu. Rev. Microbiol. 2022, 76, 21–43. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [Green Version]
- Marsan, D.; Place, A.; Fucich, D.; Chen, F. Toxin-antitoxin systems in estuarine Synechococcus strain CB0101 and their transcriptomic responses to environmental stressors. Front. Microbiol. 2017, 8, 1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucich, D.; Chen, F. Presence of toxin-antitoxin systems in picocyanobacteria and their ecological implications. ISME J. 2020, 14, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Marsan, D.; Wommack, K.E.; Ravel, J.; Chen, F. Draft Genome Sequence of Synechococcus sp. Strain CB0101, Isolated from the Chesapeake Bay Estuary. Genome Announc. 2014, 2, e01111-13. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Hazan, R.; Amitai, S. mazEF: A chromosomal toxin-antitoxin module that triggers programmed cell death in bacteria. J. Cell Sci. 2005, 118, 4327–4332. [Google Scholar] [CrossRef] [Green Version]
- Šulčius, S.; Šimoliūnas, E.; Alzbutas, G.; Gasiūnas, G.; Jauniškis, V.; Kuznecova, J.; Miettinen, S.; Nilsson, E.; Meškys, R.; Roine, E.; et al. Genomic Characterization of Cyanophage vB_AphaS-CL131 Infecting Filamentous Diazotrophic Cyanobacterium Aphanizomenon flos-aquae Reveals Novel Insights into Virus-Bacterium Interactions. Appl. Environ. Microbiol. 2019, 85, e01311-18. [Google Scholar] [CrossRef] [Green Version]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Snider, J.; Houry, W.A. MoxR AAA+ ATPases: A novel family of molecular chaperones? J. Struct. Biol. 2006, 156, 200–209. [Google Scholar] [CrossRef]
- Bakkouri, M.E.; Gutsche, I.; Kanjee, U.; Zhao, B.; Yu, M.; Goret, G.; Schoehn, G.; Burmeister, W.P.; Houry, W.A. Structure of RavA MoxR AAA+ protein reveals the design principles of a molecular cage modulating the inducible lysine decarboxylase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 22499–22504. [Google Scholar] [CrossRef] [Green Version]
- Dieppedale, J.; Sobral, D.; Dupuis, M.; Dubail, I.; Klimentova, J.; Stulik, J.; Postic, G.; Frapy, E.; Meibom, K.L.; Barel, M.; et al. Identification of a Putative Chaperone Involved in Stress Resistance and Virulence in Francisella tularensis. Infect. Immun. 2011, 79, 1428–1439. [Google Scholar] [CrossRef] [Green Version]
- Scheele, U.; Erdmann, S.; Ungewickell, E.J.; Felisberto-Rodrigues, C.; Ortiz-Lombardía, M.; Garrett, R.A. Chaperone role for proteins p618 and p892 in the extracellular tail development of Acidianus two-tailed virus. J. Virol. 2011, 85, 4812–4821. [Google Scholar] [CrossRef] [Green Version]
- Sancar, G.B. DNA repair enzymes. Annu. Rev. Biochem. 1988, 57, 29–67. [Google Scholar] [CrossRef]
- Lee, J.; Park, H.S.; Lim, S.; Jo, K. Visualization of UV-induced damage on single DNA molecules. Chem. Commun. 2013, 49, 4740–4742. [Google Scholar] [CrossRef]
- Grafstrom, R.H.; Park, L.; Grossman, L. Enzymatic repair of pyrimidine dimer-containing DNA. A 5′ dimer DNA glycosylase: 3′-apyrimidinic endonuclease mechanism from Micrococcus luteus. J. Biol. Chem. 1982, 257, 13465–13474. [Google Scholar] [CrossRef]
- Walker, R.K.; McCullough, A.K.; Lloyd, R.S. Uncoupling of Nucleotide Flipping and DNA Bending by the T4 Pyrimidine Dimer DNA Glycosylase. Biochemistry 2006, 45, 14192–14200. [Google Scholar] [CrossRef] [Green Version]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Albers, S.; Czech, A. Exploiting tRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.H. Function of the bacteriophage T4 transfer RNA’s. J. Mol. Biol. 1973, 74, 753–757. [Google Scholar] [CrossRef]
- Bobrovskyy, M.; Vanderpool, C.K. Regulation of Bacterial Metabolism by Small RNAs Using Diverse Mechanisms. Annu. Rev. Genet. 2013, 47, 209–232. [Google Scholar] [CrossRef]
- Oglesby-Sherrouse, A.G.; Murphy, E.R. Iron-responsive bacterial small RNAs: Variations on a theme. Metallomics 2013, 5, 276–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mika, F.; Hengge, R. Small RNAs in the control of RpoS, CsgD, and biofilm architecture of Escherichia coli. RNA Biol. 2014, 11, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klähn, S.; Bolay, P.; Wright, P.R.; Atilho, R.M.; Brewer, K.I.; Hagemann, M.; Breaker, R.R.; Hess, W.R. A glutamine riboswitch is a key element for the regulation of glutamine synthetase in cyanobacteria. Nucleic Acids Res. 2018, 46, 10082–10094. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Strain | Phylogenetic Clade | Isolation Source | Medium Salinity | Reference | Result a |
|---|---|---|---|---|---|
| Estuarine strains | |||||
| Synechococcus sp. CB0101 | CB4, subcluster 5.2 | Chesapeake Bay | 15 | [21] | + |
| Synechococcus sp. A10-1-5-1 | CB5, subcluster 5.2 | Changjiang River Estuary | 25 | Xu et al. unpublished | − |
| Synechococcus sp. CBW1003 | Bornholm Sea | Chesapeake Bay | 15 | [50] | − |
| Synechococcus sp. CBW1006 | Bornholm Sea | Chesapeake Bay | 15 | [50] | − |
| Synechococcus sp. CBW1107 | Subalpine C II | Chesapeake Bay | 15 | [50] | − |
| Synechococcus sp. CBW1004 | Unclassified | Chesapeake Bay | 15 | [50] | − |
| Synechococcus sp. PCC 7002 | Unclassified | Magueyes Island | 22 | [51] | − |
| Marine strains | |||||
| Synechococcus sp. CC9311 | I, subcluster 5.1 | California Current | 35 | [52] | − |
| Synechococcus sp. WH 8102 | III, subcluster 5.1 | Tropical Atlantic | 30 | [53] | − |
| Synechococcus sp. WH 7803 | V, subcluster 5.1 | Sargasso Sea | 15 | [29] | − |
| Synechococcus sp. WH 7805 | VI, subcluster 5.1 | Sargasso Sea | 35 | [29] | − |
| ORF No. | Antitoxin | TA System | TA System in the Host Genome |
|---|---|---|---|
| 54 | YefM | YefM–YoeB | + |
| 106 | TacA | TacA–TacT | − |
| 155 | MazE | MazE–MazF | + |
| Codon a | Attribute | Usage (‰) | RSCU | Codon a | Attribute | Usage (‰) | RSCU | Codon a | Attribute | Usage (‰) | RSCU | Codon a | Attribute | Usage (‰) | RSCU | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phage | Host | Phage | Host | Phage | Host | Phage | Host | Phage | Host | Phage | Host | Phage | Host | Phage | Host | ||||||||
| TTT | Phe | 18.7 | 6.3 | 0.9 | 0.7 | TCT | Ser | 22.8 | 9.2 | 1.6 | 0.6 | TAT | Tyr | 23.6 | 2.5 | 1.3 | 0.7 | TGT | Cys | 12.1 | 8.5 | 1.1 | 0.5 |
| TTC | Phe | 21.7 | 11.8 | 1.1 | 1.3 | TCC | Ser | 10.5 | 14.1 | 0.7 | 0.9 | TAC | Tyr | 13.9 | 4.2 | 0.7 | 1.3 | TGC | Cys | 10.9 | 24.4 | 1 | 1.5 |
| TTA | Leu | 17.7 | 2.1 | 1 | 0.2 | TCA | Ser | 21.6 | 15.3 | 1.5 | 1 | TAA | Stop | 17.2 | 1.9 | 1.1 | 0.3 | TGA | Stop | 19.7 | 15.3 | 1.3 | 2.2 |
| TTG | Leu | 20.8 | 12.8 | 1.2 | 1.1 | TCG | Ser | 9.5 | 17.6 | 0.7 | 1.2 | TAG | Stop | 10 | 3.5 | 0.6 | 0.5 | TGG | Trp | 17.1 | 25 | 1 | 1 |
| CTT | Leu | 18.1 | 11.3 | 1 | 0.9 | CCT | Pro | 11.6 | 18.8 | 1.4 | 0.8 | CAT | His | 13.9 | 12 | 1.2 | 0.7 | CGT | Arg | 6.1 | 11.9 | 0.6 | 0.6 |
| CTC | Leu | 11.7 | 17.5 | 0.7 | 1.4 | CCC | Pro | 4.8 | 21.9 | 0.6 | 0.9 | CAC | His | 10.1 | 21.3 | 0.8 | 1.3 | CGC | Arg | 3.2 | 31.9 | 0.3 | 1.6 |
| CTA | Leu | 19.9 | 4 | 1.1 | 0.3 | CCA | Pro | 13.3 | 27.3 | 1.6 | 1.1 | CAA | Gln | 27.6 | 14.9 | 1.2 | 0.7 | CGA | Arg | 7.7 | 19.4 | 0.8 | 1 |
| CTG | Leu | 19.8 | 29.1 | 1.1 | 2.3 | CCG | Pro | 3.1 | 27.7 | 0.4 | 1.2 | CAG | Gln | 16.8 | 29.2 | 0.8 | 1.3 | CGG | Arg | 3.1 | 27.7 | 0.3 | 1.4 |
| ATT | Ile | 26.1 | 5.5 | 1.2 | 0.6 | ACT | Thr | 24.4 | 6.3 | 1.6 | 0.6 | AAT | Asn | 24.3 | 5.5 | 1 | 0.6 | AGT | Ser | 16.7 | 5.9 | 1.1 | 0.4 |
| ATC | Ile | 23.2 | 18.5 | 1.1 | 2.1 | ACC | Thr | 10.7 | 18.4 | 0.7 | 1.6 | AAC | Asn | 22.3 | 11.5 | 1 | 1.4 | AGC | Ser | 7.3 | 27.9 | 0.5 | 1.9 |
| ATA | Ile | 16.6 | 2.5 | 0.8 | 0.3 | ACA | Thr | 18.5 | 9 | 1.2 | 0.8 | AAA | Lys | 26.9 | 6.8 | 1 | 0.7 | AGA | Arg | 26.6 | 9.6 | 2.8 | 0.5 |
| ATG | Met | 23.7 | 11.1 | 1 | 1 | ACG | Thr | 7.2 | 11.4 | 0.5 | 1 | AAG | Lys | 26.1 | 11.2 | 1 | 1.3 | AGG | Arg | 10.4 | 17.4 | 1.1 | 1 |
| GTT | Val | 20.3 | 11 | 1.3 | 1 | GCT | Ala | 12.2 | 28.6 | 1.3 | 0.9 | GAT | Asp | 26.8 | 19.3 | 1.4 | 1.4 | GGT | Gly | 23.1 | 18 | 1.8 | 0.8 |
| GTC | Val | 8.2 | 8.4 | 0.5 | 0.8 | GCC | Ala | 2.8 | 39.4 | 0.3 | 1.3 | GAC | Asp | 11 | 9.1 | 0.6 | 0.7 | GGC | Gly | 4.6 | 39.4 | 0.4 | 1.7 |
| GTA | Val | 17.8 | 4 | 1.2 | 0.4 | GCA | Ala | 17.7 | 26 | 1.9 | 0.8 | GAA | Glu | 28.6 | 12.5 | 1.3 | 0.8 | GGA | Gly | 17 | 14.7 | 1.4 | 0.6 |
| GTG | Val | 14.2 | 20.3 | 0.9 | 1.9 | GCG | Ala | 4.1 | 31.2 | 0.4 | 1 | GAG | Glu | 16.3 | 17.6 | 0.7 | 1.2 | GGG | Gly | 5.8 | 21.8 | 0.5 | 0.9 |
| Feature | Type | Strand | Start | End | Rfam Accession No. | Score | E-Value |
|---|---|---|---|---|---|---|---|
| abiF | sRNA | + | 10531 | 10567 | RF03085 | 51.6 | 3.2E-06 |
| wcaG | cis-regulatory | + | 9745 | 9842 | RF01761 | 91.4 | 2.3E-15 |
| manA | cis-regulatory | + | 10215 | 10429 | RF01745 | 100.6 | 4.6E-23 |
| glnA | cis-regulatory | − | 164330 | 164232 | RF01739 | 44.0 | 5.2E-06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Liu, Y.; Zhou, R.; Liu, J.; Xu, Y.; Chen, F. An Estuarine Cyanophage S-CREM1 Encodes Three Distinct Antitoxin Genes and a Large Number of Non-Coding RNA Genes. Viruses 2023, 15, 380. https://doi.org/10.3390/v15020380
Zheng H, Liu Y, Zhou R, Liu J, Xu Y, Chen F. An Estuarine Cyanophage S-CREM1 Encodes Three Distinct Antitoxin Genes and a Large Number of Non-Coding RNA Genes. Viruses. 2023; 15(2):380. https://doi.org/10.3390/v15020380
Chicago/Turabian StyleZheng, Hongrui, Yuanfang Liu, Ruiyu Zhou, Jihua Liu, Yongle Xu, and Feng Chen. 2023. "An Estuarine Cyanophage S-CREM1 Encodes Three Distinct Antitoxin Genes and a Large Number of Non-Coding RNA Genes" Viruses 15, no. 2: 380. https://doi.org/10.3390/v15020380
APA StyleZheng, H., Liu, Y., Zhou, R., Liu, J., Xu, Y., & Chen, F. (2023). An Estuarine Cyanophage S-CREM1 Encodes Three Distinct Antitoxin Genes and a Large Number of Non-Coding RNA Genes. Viruses, 15(2), 380. https://doi.org/10.3390/v15020380