
Molecular Characterization and Cluster Analysis of SARS-CoV-2 Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One Month

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Diagnostic RT-PCR
2.2. Sanger Sequencing
2.3. Viral Classification and Mutational Analysis
2.4. Phylogenetic and Cluster Analyses
3. Results
3.1. Patient Characteristics and ROUTINE Diagnosis
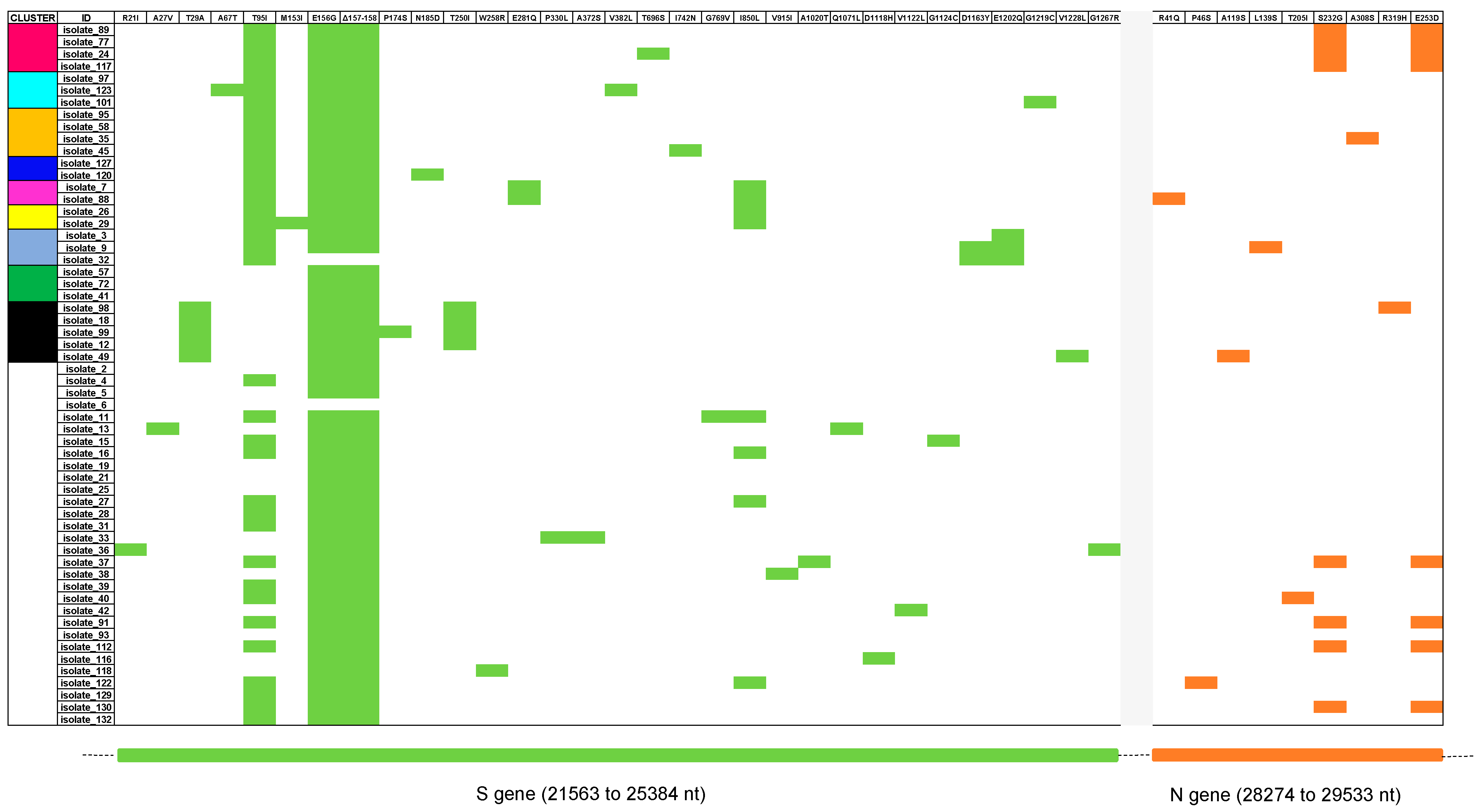
3.2. Classification and Mutation Pattern of Isolates
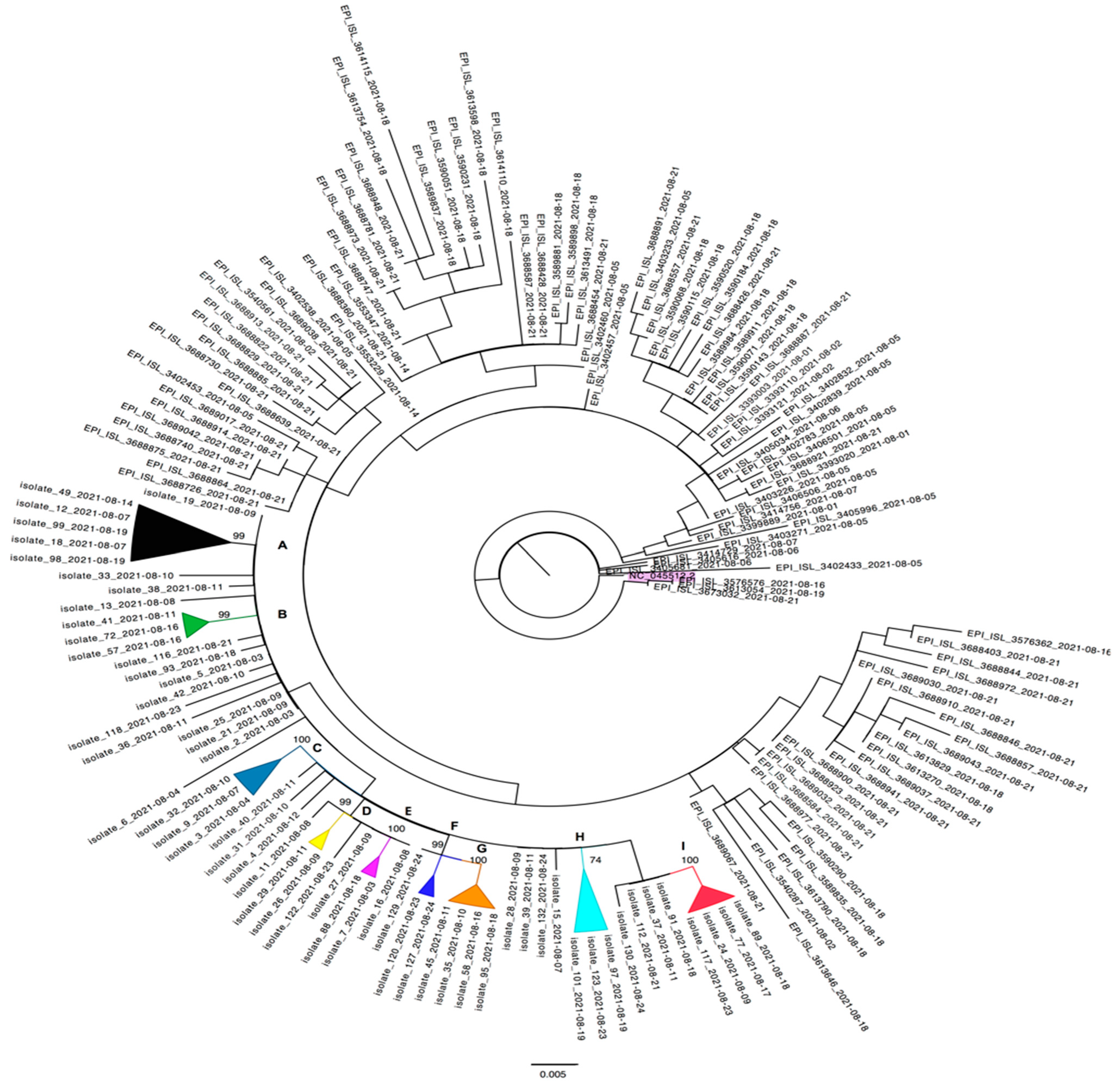
3.3. Cluster Investigation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esakandari, H.; Nabi-Afjadi, M.; Fakkari-Afjadi, J.; Farahmandian, N.; Miresmaeili, S.M.; Bahreini, E. A comprehensive review of COVID-19 characteristics. Biol. Proced. Online 2020, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Blish, C.A.; Sallusto, F.; Iwasaki, A. The immunology and immunopathology of COVID-19. Science 2022, 375, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Çağdaş, D. Living the SARS-CoV-2 pandemic in Turkey. Nat. Immunol. 2021, 22, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Bozdayi, G.; Yigit, S.; Muftah, H.; Dizbay, M.; Tunccan, O.G.; Fidan, I.; Caglar, K. Genomic characterization of SARS-CoV-2 isolates from patients in Turkey reveals the presence of novel mutations in spike and nsp12 proteins. J. Med. Virol. 2021, 93, 6016–6026. [Google Scholar] [CrossRef] [PubMed]
- Barlas, G.; Öztürk, H.; Pehlivantürk, G.; Aydin, S. Turkey’s response to COVID-19 pandemic: Strategy and key actions. Turk. J. Med. Sci. 2021, 51, 3150–3156. [Google Scholar] [CrossRef]
- Genç, K. COVID-19 in Turkey: A nation on edge. Lancet 2021, 397, 1794–1796. [Google Scholar] [CrossRef]
- Russo, A.; Serapide, F.; Quirino, A.; Tarsitano, M.G.; Marascio, N.; Serraino, R.; Rotundo, S.; Matera, G.; Trecarichi, E.M.; Torti, C. Microbiological and Clinical Findings of SARS-CoV-2 Infection after 2 Years of Pandemic: From Lung to Gut Microbiota. Diagnostics 2022, 12, 2143. [Google Scholar] [CrossRef]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 31 January 2023).
- Lim, C.; Nam, Y.; Oh, W.S.; Ham, S.; Kim, E.; Kim, M.; Kim, S.; Kim, Y.; Jeong, S. Characteristics of transmission routes of COVID-19 cluster infections in Gangwon Province, Korea. Epidemiol. Infect. 2022, 150, e19. [Google Scholar] [CrossRef]
- Safer, M.; Letaief, H.; Hechaichi, A.; Harizi, C.; Dhaouadi, S.; Bouabid, L.; Darouiche, S.; Gharbi, D.; Elmili, N.; Ben Salah, H.; et al. Identification of transmission chains and clusters associated with COVID-19 in Tunisia. BMC Infect. Dis. 2021, 21, 453. [Google Scholar] [CrossRef]
- Uzun, O.; Akpolat, T.; Varol, A.; Turan, S.; Bektas, S.G.; Cetinkaya, P.D.; Dursun, M.; Bakan, N.; Ketencioglu, B.B.; Bayrak, M.; et al. COVID-19: Vaccination vs. hospitalization. Infection 2022, 50, 747–752. [Google Scholar] [CrossRef]
- Nyberg, T.; Ferguson, N.M.; Nash, S.G.; Webster, H.H.; Flaxman, S.; Andrews, N.; HinsleY, W.; Bernal, J.L.; Kall, M.; Bhatt, S.; et al. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 omicron (B.1.1.529) and delta (B.1.617.2) variants in England: A cohort study. Lancet 2022, 399, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Hoteit, R.; Yassine, H.M. Biological Properties of SARS-CoV-2 Variants: Epidemiological Impact and Clinical Consequences. Vaccines 2022, 10, 919. [Google Scholar] [CrossRef]
- Sun, C.; Xie, C.; Bu, G.L.; Zhong, L.Y.; Zeng, M.S. Molecular characteristics, immune evasion, and impact of SARS-CoV-2 variants. Signal Transduct. Target. Ther. 2022, 7, 202. [Google Scholar] [CrossRef]
- Ganesh, B.; Rajakumar, T.; Malathi, M.; Manikandan, N.; Nagaraj, J.; Santhakumar, A.; Elangovan, A.; Malik, Y.S. Epidemiology and pathobiology of SARS-CoV-2 (COVID-19) in comparison with SARS, MERS: An updated overview of current knowledge and future perspectives. Clin. Epidemiol. Glob. Health 2021, 10, 100694. [Google Scholar] [CrossRef] [PubMed]
- Eskİer, D.; Akalp, E.; Dalan, O.; Ülah, G.K.; Oktay, Y. Current mutatome of SARS-CoV-2 in Turkey reveals mutations of interest. Turk. J. Biol. 2021, 45, 104–113. [Google Scholar] [CrossRef]
- Ng, O.H.; Akyoney, S.; Sahin, I.; Soykam, H.O.; Akcapinar, G.B.; Ozdemir, O.; Kancagi, D.D.; Karakus, G.S.; Yurtsever, B.; Kocagoz, A.S.; et al. Mutational landscape of SARS-CoV-2 genome in Turkey and impact of mutations on spike protein structure. PLoS ONE 2021, 16, e0260438. [Google Scholar] [CrossRef]
- Ergünay, K.; Kaya, M.; Serdar, M.; Akyön, Y.; Yılmaz, E. A cross-sectional overview of SARS-CoV-2 genome variations in Turkey. Turk. J. Biochem. 2021, 46, 491–498. [Google Scholar] [CrossRef]
- Mohammad, T.; Choudhury, A.; Habib, I.; Asrani, P.; Mathur, Y.; Umair, M.; Anjum, F.; Shafie, A.; Yadav, D.K.; Hassan, M.I. Genomic Variations in the Structural Proteins of SARS-CoV-2 and Their Deleterious Impact on Pathogenesis: A Comparative Genomics Approach. Front. Cell. Infect. Microbiol. 2021, 11, 765039. [Google Scholar] [CrossRef]
- Rahman, M.S.; Islam, M.R.; Alam, A.; Islam, I.; Hoque, M.N.; Akter, S.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Evolutionary dynamics of SARS-CoV-2 nucleocapsid protein and its consequences. J. Med. Virol. 2021, 93, 2177–2195. [Google Scholar] [CrossRef]
- Zhao, H.; Nguyen, A.; Wu, D.; Li, Y.; Hassan, S.A.; Chen, J.; Shroff, H.; Piszczek, G.; Schuck, P. Plasticity in structure and assembly of SARS-CoV-2 nucleocapsid protein. PNAS Nexus. 2022, 1, pgac049. [Google Scholar] [CrossRef]
- Alkhatib, M.; Bellocchi, M.C.; Marchegiani, G.; Grelli, S.; Micheli, V.; Stella, D.; Zerillo, B.; Carioti, L.; Svicher, V.; Rogliani, P.; et al. First Case of a COVID-19 Patient Infected by Delta AY.4 with a Rare Deletion Leading to a N Gene Target Failure by a Specific Real Time PCR Assay: Novel Omicron VOC Might Be Doing Similar Scenario? Microorganisms 2022, 10, 268. [Google Scholar] [CrossRef]
- Paden, C.R.; Tao, Y.; Queen, K.; Zhang, J.; Li, Y.; Uehara, A.; Tong, S. Rapid, Sensitive, Full-Genome Sequencing of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 2401–2405. [Google Scholar] [CrossRef] [PubMed]
- Pangolin COVID-19 Lineage Assigner. Available online: https://pangolin.cog-uk.io (accessed on 20 October 2022).
- GISAID-CoVsurver Mutations App. Available online: https://www.gisaid.org/epiflu-applications/covsurver-mutations-app/ (accessed on 23 October 2022).
- Stanford Coronavirus Resistance Database CoV-RDB. Available online: https://covdb.stanford.edu (accessed on 25 October 2022).
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2014, 42, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Kuiken, C.; Hraber, P.; Thurmond, J.; Yusim, K. The hepatitis C sequence database in Los Alamos. Nucleic Acids Res. 2008, 36, 512–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucl. Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B. 1.617. 2) variant in vaccinated and unvaccinated individuals in the UK: A prospective, longitudinal, cohort study. Lancet Infect. Dis. 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Benda, A.; Zerajic, L.; Ankita, A.; Cleary, E.; Park, Y.; Pandey, S. COVID-19 Testing and Diagnostics: A Review of Commercialized Technologies for Cost, Convenience and Quality of Tests. Sensors 2021, 21, 6581. [Google Scholar] [CrossRef] [PubMed]
- Guney, M.; Hosbul, T.; Cuce, F.; Artuk, C.; Taskin, G.; Caglayan, M.; Alacam, S.; Kurkcu, M.F.; Yildiz, F.; Erdal, H.; et al. Evaluation of the relationship between progression and SARS-CoV-2 viral load in COVID-19 cases in Ankara, Turkey. J. Infect. Dev. Ctries. 2022, 16, 462–468. [Google Scholar] [CrossRef]
- Yusof, W.; Irekeola, A.A.; Wada, Y.; Engku Abd Rahman, E.N.S.; Ahmed, N.; Musa, N.; Khalid, M.F.; Rahman, Z.A.; Hassan, R.; Yusof, N.Y.; et al. A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients. Life 2021, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- De Marco, C.; Marascio, N.; Veneziano, C.; Biamonte, F.; Trecarichi, E.M.; Santamaria, G.; Leviyang, S.; Liberto, M.C.; Mazzitelli, M.; Quirino, A.; et al. Whole-genome analysis of SARS-CoV-2 in a 2020 infection cluster in a nursing home of Southern Italy. Infect. Genet. Evol. 2022, 99, 105253. [Google Scholar] [CrossRef]
- Leducq, V.; Couturier, J.; Granger, B.; Jolivet, S.; Morand-Joubert, L.; Robert, J.; Denis, M.; Salauze, B.; Goldstein, V.; Zafilaza, K.; et al. Investigation of healthcare-associated COVID-19 in a large French hospital group by whole-genome sequencing. Microbiol. Res. 2022, 263, 127133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Asghar, A.; Raza, K.; Narayan, R.K.; Jha, R.K.; Satyam, A.; Kumar, G.; Dwivedi, P.; Sahni, C.; Kumari, C. Demographic characteristics of SARS-CoV-2 B. 1.617. 2 (Delta) variant infections in Indian population. medRxiv, 2021; Preprint. [Google Scholar] [CrossRef]
- Guruprasad, K. Mutations in human SARS-CoV-2 spike proteins, potential drug binding and epitope sites for COVID-19 therapeutics development. Curr. Res. Struct. Biol. 2022, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.; Pyle, J.; Yurkovetskiy, L.; Bosso, M.; Park, D.; Babadi, M.; MacInnis, B.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef]
- Rajpal, V.R.; Sharma, S.; Kumar, A.; Vaishnavi, S.; Singh, A.; Sehgal, D.; Tiwari, M.; Goel, S.; Raina, S.N. Mapping of SARS-CoV-2 spike protein evolution during first and second waves of COVID-19 infections in India. Future Virol. 2022, 17, 557–575. [Google Scholar] [CrossRef]
- Trigueiro-Louro, J.; Correia, V.; Figueiredo-Nunes, I.; Gíria, M.; Rebelo-de-Andrade, H. Unlocking COVID therapeutic targets: A structure-based rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Comput. Struct. Biotechnol. J. 2020, 18, 2117–2131. [Google Scholar] [CrossRef]
- McBride, R.; Zyl, M.; Fielding, B.C. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Nalla, A.K.; Casto, A.M.; Huang, M.W.; Perchetti, G.A.; Sampoleo, R.; Shrestha, L.; Wei, Y.; Zhu, H.; Jerome, K.R.; Greninger, A.L. Comparative Performance of SARS-CoV-2 Detection Assays Using Seven Different Primer-Probe Sets and One Assay Kit. J. Clin. Microbiol. 2020, 58, e00557-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasetyoputri, A.; Dharmayanthi, A.B.; Iryanto, S.B.; Andriani, A.; Nuryana, I.; Wardiana, A.; Ridwanuloh, A.M.; Swasthikawati, S.; Hariyatun, H.; Nugroho, H.A.; et al. The dynamics of circulating SARS-CoV-2 lineages in Bogor and surrounding areas reflect variant shifting during the first and second waves of COVID-19 in Indonesia. PeerJ 2022, 10, e13132. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, A.; Kovalenko, G.; Redlinger, M.; Liulchuk, M.G.; Bortz, E.; Zadorozhna, V.I.; Scherbinska, A.M.; Wertheim, J.O.; Goodfellow, I.; Meredith, L.; et al. Tracking SARS-COV-2 variants using Nanopore sequencing in Ukraine in 2021. Sci. Rep. 2022, 12, 15749. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Total (%) | Male (%) | Female (%) | ||||
|---|---|---|---|---|---|---|
| Number of Cases | 58 | 100 | 27 | 46.6 | 31 | 53.4 |
| Age (Years-Old) | ||||||
| 0–9 | 1 | 1.7 | 1 | 100.0 | 0 | 0.0 |
| 10–19 | 14 | 24.1 | 6 | 42.9 | 8 | 57.1 |
| 20–29 | 9 | 15,5 | 5 | 55.6 | 4 | 44.4 |
| 30–39 | 14 | 24.1 | 7 | 50.0 | 7 | 50.0 |
| 40–49 | 10 | 17.2 | 5 | 50.0 | 5 | 50.0 |
| 50–59 | 3 | 5.2 | 1 | 33.3 | 2 | 66.7 |
| 60–69 | 5 | 8.6 | 1 | 20.0 | 4 | 80.0 |
| ≥70 | 2 | 3.4 | 1 | 50.0 | 1 | 50.0 |
| Clinical manifestations * | ||||||
| Aspecific symptoms | 2 | 3.4 | 0 | 0.0 | 2 | 100.0 |
| Backache | 1 | 1.7 | 0 | 0.0 | 1 | 100.0 |
| Acute pain + malaise + fatigue | 1 | 1.7 | 0 | 0.0 | 1 | 100.0 |
| COVID-19 symptoms | 34 | 58.6 | 18 | 52.9 | 16 | 47.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marascio, N.; Cilburunoglu, M.; Torun, E.G.; Centofanti, F.; Mataj, E.; Equestre, M.; Bruni, R.; Quirino, A.; Matera, G.; Ciccaglione, A.R.; et al. Molecular Characterization and Cluster Analysis of SARS-CoV-2 Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One Month. Viruses 2023, 15, 802. https://doi.org/10.3390/v15030802
Marascio N, Cilburunoglu M, Torun EG, Centofanti F, Mataj E, Equestre M, Bruni R, Quirino A, Matera G, Ciccaglione AR, et al. Molecular Characterization and Cluster Analysis of SARS-CoV-2 Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One Month. Viruses. 2023; 15(3):802. https://doi.org/10.3390/v15030802
Chicago/Turabian StyleMarascio, Nadia, Merve Cilburunoglu, Elif Gulsum Torun, Federica Centofanti, Elida Mataj, Michele Equestre, Roberto Bruni, Angela Quirino, Giovanni Matera, Anna Rita Ciccaglione, and et al. 2023. "Molecular Characterization and Cluster Analysis of SARS-CoV-2 Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One Month" Viruses 15, no. 3: 802. https://doi.org/10.3390/v15030802
APA StyleMarascio, N., Cilburunoglu, M., Torun, E. G., Centofanti, F., Mataj, E., Equestre, M., Bruni, R., Quirino, A., Matera, G., Ciccaglione, A. R., & Yalcinkaya, K. T. (2023). Molecular Characterization and Cluster Analysis of SARS-CoV-2 Viral Isolates in Kahramanmaraş City, Turkey: The Delta VOC Wave within One Month. Viruses, 15(3), 802. https://doi.org/10.3390/v15030802