
Stabilization of the Quadruplex-Forming G-Rich Sequences in the Rhinovirus Genome Inhibits Uncoating—Role of Na+ and K+

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis of Rhinoviral Genomes for Identification of Putative GQs
2.2. Oligonucleotides and QG-Binding Compounds
2.3. Cells and Virus
2.4. ThT Assay for Detection of GQ
2.5. Fluorescent Indicator Displacement Assay (FiD)
2.6. The Particle Stability Thermal Release Assay (PaSTRy)
2.7. Melting Analysis of RV-A2 ‘Ex-Virion’ RNA
2.8. Virus Yield Assay
2.9. Immunocytochemistry and Flow Cytometry
2.10. Sucrose Density Gradient Sedimentation
2.11. Time-of-Drug-Addition Experiments
2.12. Electron Microscopy and Rotary Shadowing
2.13. Atomic Force Microscopy (AFM)
2.14. Quantification of PDS Binding to the Virus
2.15. Quantification and Statistical Analysis
3. Results
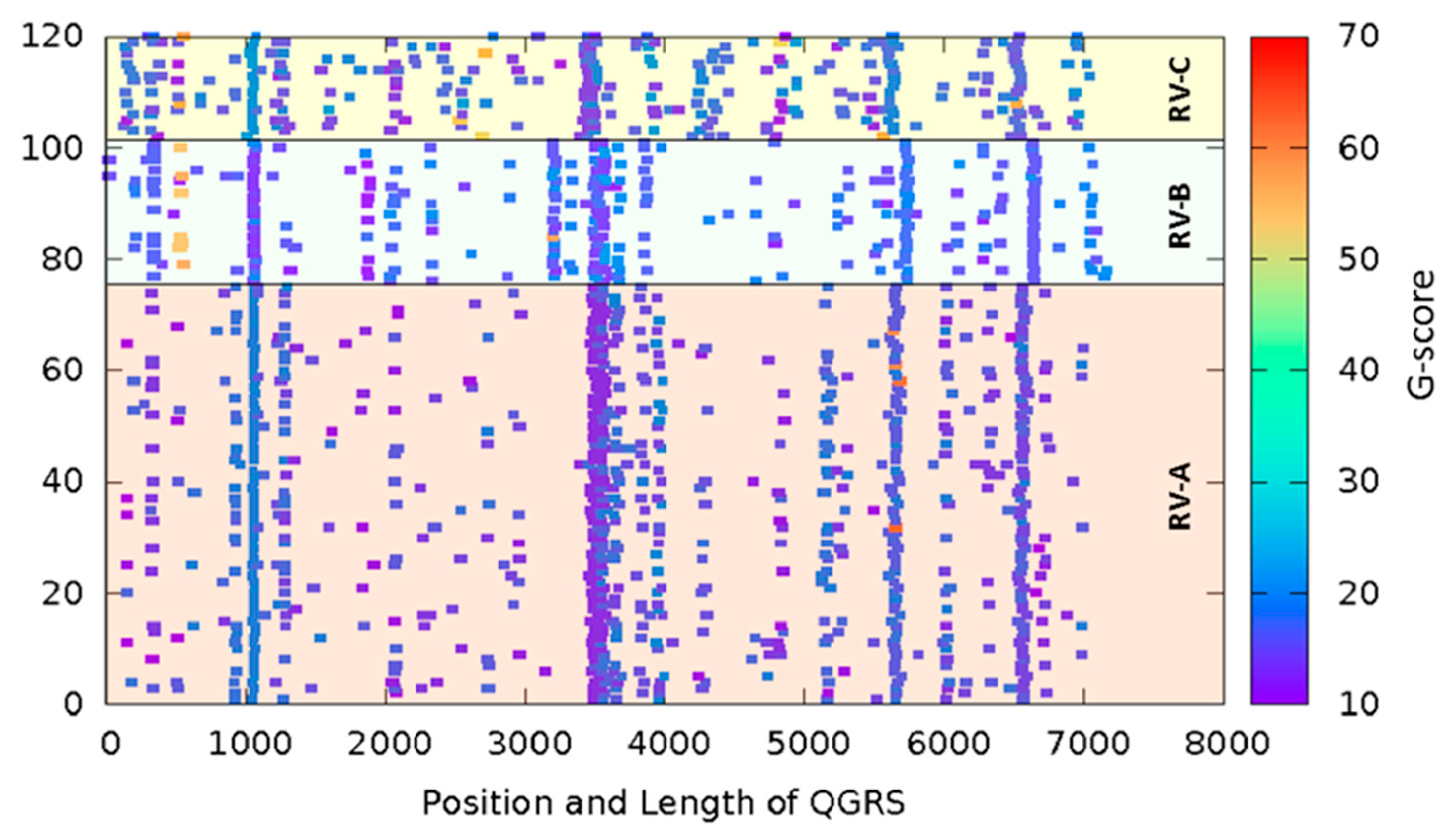
3.1. QGRS Mapper Identifies Potential GQ-Forming Sequences in All RV Genomes
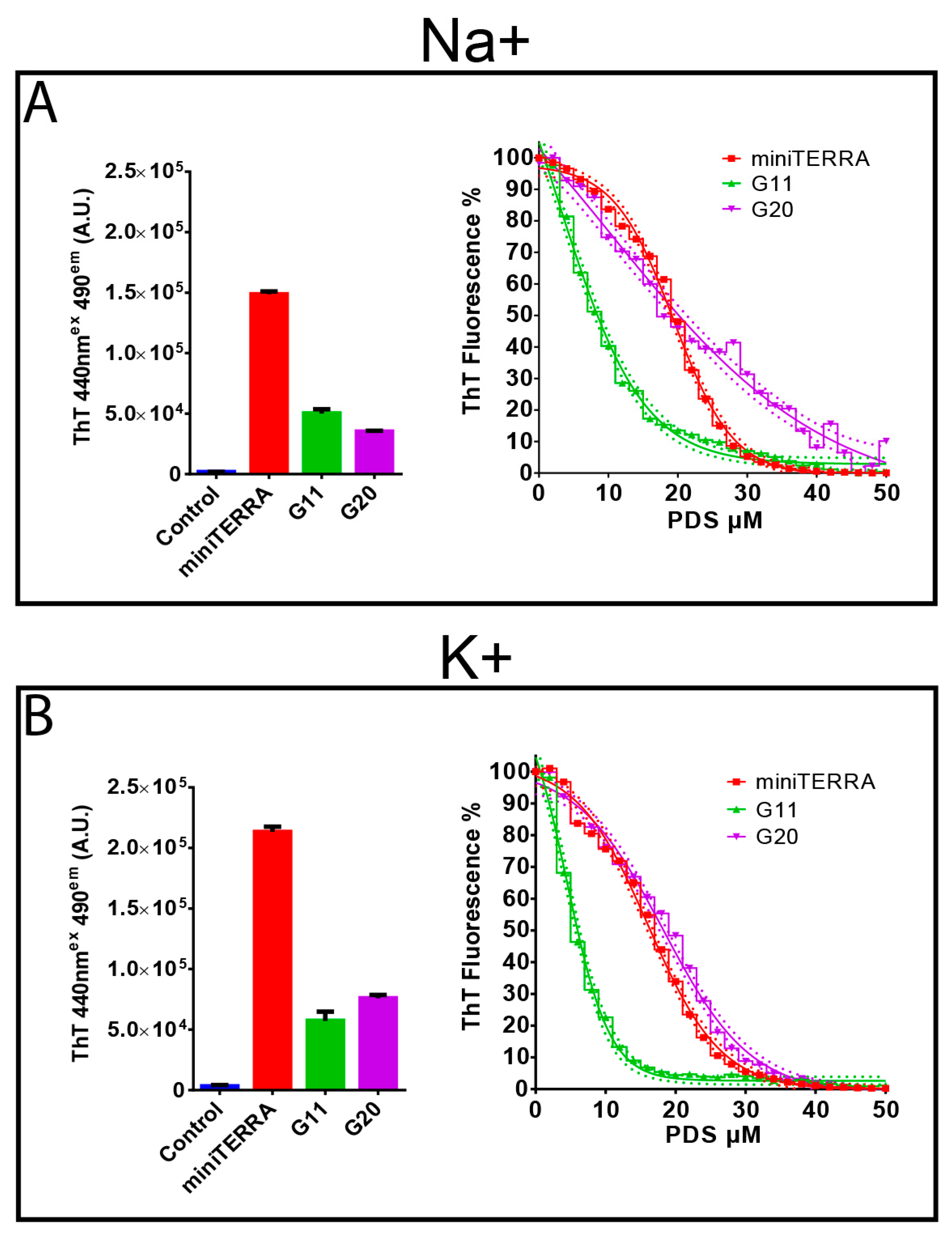
3.2. Thioflavin T Light-Up Assay and Competition with PDS Demonstrate Specific Binding
3.3. PDS and PhenDC3 Reduce RV-A2 Infectivity
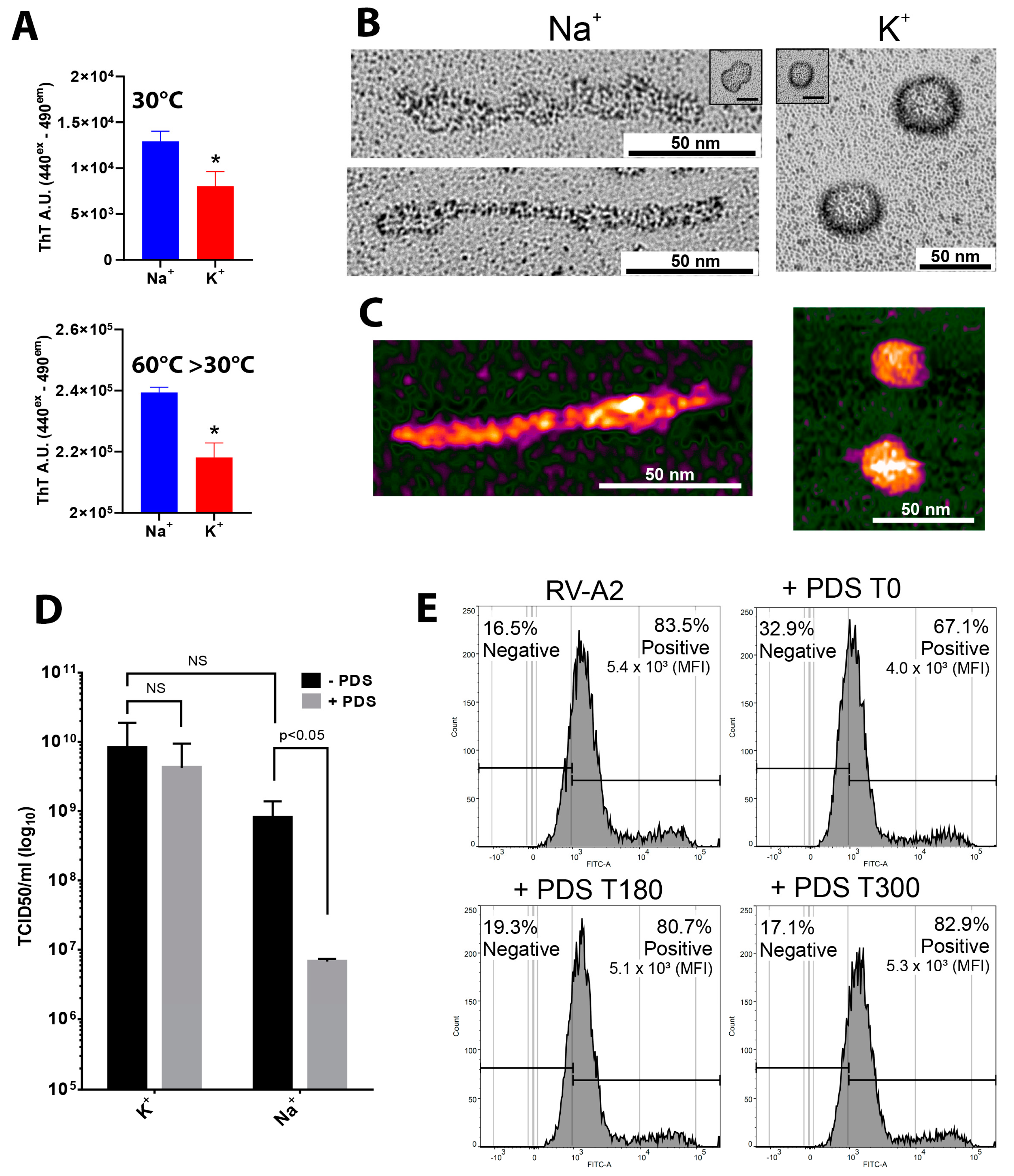
3.4. PDS Affects the Conformation of RV-A2 Genomic RNA and Reduces Viral Infectivity in the Presence of Na+ but Not K+
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esneau, C.; Duff, A.C.; Bartlett, N.W. Understanding Rhinovirus Circulation and Impact on Illness. Viruses 2022, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef]
- Gern, J.E.; Busse, W.W. Association of Rhinovirus Infections with Asthma. Clin. Microbiol. Rev. 1999, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Lam, T.T.; Zaraket, H.; Lipkin, W.I.; Drews, S.J.; Hatchette, T.F.; Heraud, J.-M.; Koopmans, M.P.; Abraham, A.M.; Baraket, A.; et al. Global epidemiology of non-influenza RNA respiratory viruses: Data gaps and a growing need for surveillance. Lancet Infect. Dis. 2017, 17, e320–e326. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Jarvinen, A. The common cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef]
- Basnet, S.; Palmenberg, A.C.; Gern, J.E. Rhinoviruses and Their Receptors. Chest 2019, 155, 1018–1025. [Google Scholar] [CrossRef]
- Pickl-Herk, A.; Luque, D.; Vives-Adrián, L.; Querol-Audí, J.; Garriga, D.; Trus, B.L.; Verdaguer, N.; Blaas, D.; Castón, J.R. Uncoating of common cold virus is preceded by RNA switching as determined by X-ray and cryo-EM analyses of the subviral A-particle. Proc. Natl. Acad. Sci. USA 2013, 110, 20063–20068. [Google Scholar] [CrossRef]
- Garriga, D.; Pickl-Herk, A.; Luque, D.; Wruss, J.; Caston, J.; Blaas, D.; Verdaguer, N. Insights into Minor Group Rhinovirus Uncoating: The X-ray Structure of the HRV2 Empty Capsid. PLoS Pathog. 2012, 8, e1002473. [Google Scholar] [CrossRef] [PubMed]
- Bostina, M.; Levy, H.; Filman, D.J.; Hogle, J.M. Poliovirus RNA Is Released from the Capsid near a Twofold Symmetry Axis. J. Virol. 2011, 85, 776–783. [Google Scholar] [CrossRef]
- Shingler, K.L.; Yoder, J.L.; Carnegie, M.S.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Hafenstein, S. The enterovirus 71 A-particle forms a gateway to allow genome release: A cryoEM study of picornavirus uncoating. PLoS Pathog. 2013, 9, e1003240. [Google Scholar] [CrossRef]
- Harutyunyan, S.; Kowalski, H.; Blaas, D. The Rhinovirus Subviral A-Particle Exposes 3′-Terminal Sequences of Its Genomic RNA. J. Virol. 2014, 88, 6307–6317. [Google Scholar] [CrossRef]
- Harutyunyan, S.; Kumar, M.; Sedivy, A.; Subirats, X.; Kowalski, H.; Köhler, G.; Blaas, D. Viral Uncoating Is Directional: Exit of the Genomic RNA in a Common Cold Virus Starts with the Poly-(A) Tail at the 3′-End. PLoS Pathog. 2013, 9, e1003270. [Google Scholar] [CrossRef]
- Kumar, M.; Blaas, D. Human Rhinovirus Subviral A Particle Binds to Lipid Membranes over a Twofold Axis of Icosahedral Symmetry. J. Virol. 2013, 87, 11309–11312. [Google Scholar] [CrossRef]
- Groppelli, E.; Levy, H.C.; Sun, E.; Strauss, M.; Nicol, C.; Gold, S.; Zhuang, X.; Tuthill, T.J.; Hogle, J.M.; Rowlands, D.J. Picornavirus RNA is protected from cleavage by ribonuclease during virion uncoating and transfer across cellular and model membranes. PLoS Pathog. 2017, 13, e1006197. [Google Scholar] [CrossRef]
- Strauss, M.; Levy, H.C.; Bostina, M.; Filman, D.J.; Hogle, J.M. RNA Transfer from Poliovirus 135S Particles across Membranes Is Mediated by Long Umbilical Connectors. J. Virol. 2013, 87, 3903–3914. [Google Scholar] [CrossRef]
- Buchta, D.; Füzik, T.; Hrebík, D.; Levdansky, Y.; Sukeník, L.; Mukhamedova, L.; Moravcová, J.; Vácha, R.; Plevka, P. Enterovirus particles expel capsid pentamers to enable genome release. Nat. Commun. 2019, 10, 1138. [Google Scholar] [CrossRef]
- Hrebík, D.; Füzik, T.; Gondová, M.; Šmerdová, L.; Adamopoulos, A.; Šedo, O.; Zdráhal, Z.; Plevka, P. ICAM-1 induced rearrangements of capsid and genome prime rhinovirus 14 for activation and uncoating. Proc. Natl. Acad. Sci. USA 2021, 118, e2024251118. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef]
- Egorova, A.; Ekins, S.; Schmidtke, M.; Makarov, V. Back to the future: Advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. [Google Scholar] [CrossRef]
- Wald, J.; Pasin, M.; Richter, M.; Walther, C.; Mathai, N.; Kirchmair, J.; Makarov, V.A.; Goessweiner-Mohr, N.; Marlovits, T.C.; Zanella, I.; et al. Cryo-EM structure of pleconaril-resistant rhinovirus-B5 complexed to the antiviral OBR-5-340 reveals unexpected binding site. Proc. Natl. Acad. Sci. USA 2019, 116, 19109–19115. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Geraets, J.A.; Ma, Y.; Mirabelli, C.; Flatt, J.W.; Domanska, A.; Delang, L.; Jochmans, D.; Kumar, T.A.; Jayaprakash, V.; et al. A novel druggable interprotomer pocket in the capsid of rhino- and enteroviruses. PLoS Biol. 2019, 17, e3000281. [Google Scholar] [CrossRef]
- Wali, L.; Karbiener, M.; Chou, S.; Kovtunyk, V.; Adonyi, A.; Gösler, I.; Contreras, X.; Stoeva, D.; Blaas, D.; Stöckl, J.; et al. Host-directed therapy with 2-deoxy-D-glucose inhibits human rhinoviruses, endemic coronaviruses, and SARS-CoV-2. J. Virus Erad. 2022, 8, 100305. [Google Scholar] [CrossRef]
- Berzal-Herranz, A.; Romero-López, C.; Berzal-Herranz, B.; Ramos-Lorente, S. Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target. Pharmaceuticals 2019, 12, 38. [Google Scholar] [CrossRef]
- Xiao, C.-D.; Shibata, T.; Yamamoto, Y.; Xu, Y. An intramolecular antiparallel G-quadruplex formed by human telomere RNA. Chem. Commun. 2018, 54, 3944–3946. [Google Scholar] [CrossRef]
- Jeng, S.C.; Chan, H.H.; Booy, E.P.; McKenna, S.A.; Unrau, P.J. Fluorophore ligand binding and complex stabilization of the RNA Mango and RNA Spinach aptamers. RNA 2016, 22, 1884–1892. [Google Scholar] [CrossRef]
- Dolinnaya, N.G.; Ogloblina, A.M.; Yakubovskaya, M.G. Structure, properties, and biological relevance of the DNA and RNA G-quadruplexes: Overview 50 years after their discovery. Biochemistry 2016, 81, 1602–1649. [Google Scholar] [CrossRef]
- Masai, H.; Tanaka, T. G-quadruplex DNA and RNA: Their roles in regulation of DNA replication and other biological functions. Biochem. Biophys. Res. Commun. 2020, 531, 25–38. [Google Scholar] [CrossRef]
- Ishiguro, A.; Kimura, N.; Watanabe, Y.; Watanabe, S.; Ishihama, A. TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells 2016, 21, 466–481. [Google Scholar] [CrossRef]
- Majee, P.; Pattnaik, A.; Sahoo, B.R.; Shankar, U.; Pattnaik, A.K.; Kumar, A.; Nayak, D. Inhibition of Zika virus replication by G-quadruplex-binding ligands. Mol. Ther. Nucleic Acids 2021, 23, 691–701. [Google Scholar] [CrossRef]
- Artusi, S.; Ruggiero, E.; Nadai, M.; Tosoni, B.; Perrone, R.; Ferino, A.; Zanin, I.; Xodo, L.; Flamand, L.; Richter, S.N. Antiviral Activity of the G-Quadruplex Ligand TMPyP4 against Herpes Simplex Virus-1. Viruses 2021, 13, 196. [Google Scholar] [CrossRef]
- Zhao, C.; Qin, G.; Niu, J.; Wang, Z.; Wang, C.; Ren, J.; Qu, X. Targeting RNA G-Quadruplex in SARS-CoV-2: A Promising Therapeutic Target for COVID-19? Angew. Chem. Int. Ed. Engl. 2021, 60, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-R.; Zhang, Q.-Y.; Wang, J.-Q.; Ge, X.-Y.; Song, Y.-Y.; Wang, Y.-F.; Li, X.-D.; Fu, B.-S.; Xu, G.-H.; Shu, B.; et al. Chemical Targeting of a G-Quadruplex RNA in the Ebola Virus L Gene. Cell Chem. Biol. 2016, 23, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Perrone, R.; Artusi, S.; Butovskaya, E.; Nadai, M.; Pannecouque, C.; Richter, S.N. G-Quadruplexes in the Human Immunodeficiency Virus-1 and Herpes Simplex Virus-1: New Targets for Antiviral Activity by Small Molecules; Springer International Publishing: Cham, Switzerland, 2015; pp. 207–210. [Google Scholar] [CrossRef]
- Brázda, V.; Porubiaková, O.; Cantara, A.; Bohálová, N.; Coufal, J.; Bartas, M.; Fojta, M.; Mergny, J.-L. G-quadruplexes in H1N1 influenza genomes. BMC Genom. 2021, 22, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-R.; Min, Y.-Q.; Wang, J.-Q.; Liu, C.-X.; Fu, B.-S.; Wu, F.; Wu, L.-Y.; Qiao, Z.-X.; Song, Y.-Y.; Xu, G.-H.; et al. A highly conserved G-rich consensus sequence in hepatitis C virus core gene represents a new anti–hepatitis C target. Sci. Adv. 2016, 2, e1501535. [Google Scholar] [CrossRef]
- Tluckova, K.; Marušič, M.; Tóthová, P.; Bauer, L.; Šket, P.; Plavec, J.; Viglasky, V. Human Papillomavirus G-Quadruplexes. Biochemistry 2013, 52, 7207–7216. [Google Scholar] [CrossRef]
- Ruggiero, E.; Richter, S.N. Viral G-quadruplexes: New frontiers in virus pathogenesis and antiviral therapy. Annu. Rep. Med. Chem. 2020, 54, 101–131. [Google Scholar] [CrossRef]
- Ruggiero, E.; Richter, S.N. G-quadruplexes and G-quadruplex ligands: Targets and tools in antiviral therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef]
- Lavezzo, E.; Berselli, M.; Frasson, I.; Perrone, R.; Palù, G.; Brazzale, A.R.; Richter, S.N.; Toppo, S. G-quadruplex forming sequences in the genome of all known human viruses: A comprehensive guide. PLoS Comput. Biol. 2018, 14, e1006675. [Google Scholar] [CrossRef]
- Lv, L.; Zhang, L. Characterization of G-Quadruplexes in Enterovirus A71 Genome and Their Interaction with G-Quadruplex Ligands. Microbiol. Spectr. 2022, 10, e0046022. [Google Scholar] [CrossRef]
- Binas, O.; Bessi, I.; Schwalbe, H. Structure Validation of G-Rich RNAs in Noncoding Regions of the Human Genome. ChemBioChem 2020, 21, 1656–1663. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Alenko, A.; Burrows, C.J. Zika Virus Genomic RNA Possesses Conserved G-Quadruplexes Characteristic of the Flaviviridae Family. ACS Infect. Dis. 2016, 2, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D'Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hagen, T.; Clery, A.; Allain, F.H.; Hall, J. Control of the polyamine biosynthesis pathway by G2-quadruplexes. eLife 2018, 7, e36362. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Cui, X.; Adrian, M.; Samazan, F.; Heddi, B.; Phan, A.T.; Nicolas, A.G. Non-Canonical G-quadruplexes cause the hCEB1 minisatellite instability in Saccharomyces cerevisiae. eLife 2017, 6, e26884. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, K.; Seimiya, H. Telomeric repeat-containing NA/G-quadruplex-forming sequences cause genome-wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015, 43, 2022–2032. [Google Scholar] [CrossRef]
- Weiss, V.U.; Subirats, X.; Kumar, M.; Harutyunyan, S.; Gösler, I.; Kowalski, H.; Blaas, D. Capillary Electrophoresis, Gas-Phase Electrophoretic Mobility Molecular Analysis, and Electron Microscopy: Effective Tools for Quality Assessment and Basic Rhinovirus Research. Rhinoviruses Methods Protoc. 2014, 1221, 101–128. [Google Scholar] [CrossRef]
- Walter, T.S.; Ren, J.; Tuthill, T.J.; Rowlands, D.J.; Stuart, D.I.; Fry, E.E. A plate-based high-throughput assay for virus stability and vaccine formulation. J. Virol. Methods 2012, 185, 166–170. [Google Scholar] [CrossRef]
- Real-Hohn, A.; Groznica, M.; Löffler, N.; Blaas, D.; Kowalski, H. nanoDSF: In vitro Label-Free Method to Monitor Picornavirus Uncoating and Test Compounds Affecting Particle Stability. Front. Microbiol. 2020, 11, 1442. [Google Scholar] [CrossRef]
- Blake, K.; O’Connell, S. Virus Culture; Blackwell Scientific Publications: London, UK, 1993; p. 41. [Google Scholar]
- Skern, T.; Neubauer, C.; Frasel, L.; Gründler, P.; Sommergruber, W.; Zorn, M.; Kuechler, E.; Blaas, D. A Neutralizing Epitope on Human Rhinovirus Type 2 Includes Amino Acid Residues between 153 and 164 of Virus Capsid Protein VP2. J. Gen. Virol. 1987, 68, 315–323. [Google Scholar] [CrossRef]
- Kremser, L.; Petsch, M.; Blaas, D.; Kenndler, E. Capillary electrophoresis of affinity complexes between subviral 80S particles of human rhinovirus and monoclonal antibody 2G2. Electrophoresis 2006, 27, 2630–2637. [Google Scholar] [CrossRef]
- Ramljak, I.C.; Stanger, J.; Real-Hohn, A.; Dreier, D.; Wimmer, L.; Redlberger-Fritz, M.; Fischl, W.; Klingel, K.; Mihovilovic, M.D.; Blaas, D.; et al. Cellular N-myristoyltransferases play a crucial picornavirus genus-specific role in viral assembly, virion maturation, and infectivity. PLoS Pathog. 2018, 14, e1007203. [Google Scholar] [CrossRef]
- Sherman, M.B.; Smith, H.Q.; Smith, T.J. The Dynamic Life of Virus Capsids. Viruses 2020, 12, 618. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.K.; Bothner, B.; Smith, T.J.; Siuzdak, G. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. USA 1998, 95, 6774–6778. [Google Scholar] [CrossRef]
- Joachimi, A.; Benz, A.; Hartig, J.S. A comparison of DNA and RNA quadruplex structures and stabilities. Bioorganic Med. Chem. 2009, 17, 6811–6815. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, Q.; Xiang, J.; Yang, Q.; Sun, H.; Guan, A.; Wang, L.; Liu, Y.; Yu, L.; Shi, Y.; et al. Thioflavin T as an efficient fluorescence sensor for selective recognition of RNA G-quadruplexes. Sci. Rep. 2016, 6, 24793. [Google Scholar] [CrossRef]
- Mestre-Fos, S.; Penev, P.I.; Richards, J.C.; Dean, W.L.; Gray, R.D.; Chaires, J.B.; Williams, L.D. Profusion of G-quadruplexes on both subunits of metazoan ribosomes. PLoS ONE 2019, 14, e0226177. [Google Scholar] [CrossRef]
- Brandenburg, B.; Lee, L.Y.; Lakadamyali, M.; Rust, M.J.; Zhuang, X.; Hogle, J.M. Imaging Poliovirus Entry in Live Cells. PLoS Biol. 2007, 5, e183. [Google Scholar] [CrossRef]
- Katpally, U.; Smith, T.J. Pocket Factors Are Unlikely To Play a Major Role in the Life Cycle of Human Rhinovirus. J. Virol. 2007, 81, 6307–6315. [Google Scholar] [CrossRef]
- Killington, R.A.; Stott, E.J.; Lee, D. The Effect of Temperature on the Synthesis of Rhinovirus Type 2 RNA. J. Gen. Virol. 1977, 36, 403–411. [Google Scholar] [CrossRef]
- Reisdorph, N.; Thomas, J.J.; Katpally, U.; Chase, E.; Harris, K.; Siuzdak, G.; Smith, T.J. Human rhinovirus capsid dynamics is controlled by canyon flexibility. Virology 2003, 314, 34–44. [Google Scholar] [CrossRef]
- Lonberg-Holm, K.; Yin, F.H. Antigenic determinants of infective and inactivated human rhinovirus type 2. J. Virol. 1973, 12, 114–123. [Google Scholar] [CrossRef]
- Hewat, E.A.; Blaas, D. Nonneutralizing Human Rhinovirus Serotype 2-Specific Monoclonal Antibody 2G2 Attaches to the Region That Undergoes the Most Dramatic Changes upon Release of the Viral RNA. J. Virol. 2006, 80, 12398–12401. [Google Scholar] [CrossRef] [PubMed]
- Real-Hohn, A.; Zhu, R.; Ganjian, H.; Ibrahim, N.; Hinterdorfer, P.; Kowalski, H.; Blaas, D. Catching Common Cold Virus with a Net: Pyridostatin Forms Filaments in Tris Buffer That Trap Viruses—A Novel Antiviral Strategy? Viruses 2020, 12, 723. [Google Scholar] [CrossRef] [PubMed]
- Lonberg-Holm, K.; Noble-Harvey, J. Comparison of in vitro and cell-mediated alteration of a human Rhinovirus and its inhibition by sodium dodecyl sulfate. J. Virol. 1973, 12, 819–826. [Google Scholar] [CrossRef]
- Piazza, A.; Boulé, J.-B.; Lopes, J.; Mingo, K.; Largy, E.; Teulade-Fichou, M.-P.; Nicolas, A. Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 2010, 38, 4337–4348. [Google Scholar] [CrossRef] [PubMed]
- Speir, J.A.; Johnson, J.E. Nucleic acid packaging in viruses. Curr. Opin. Struct. Biol. 2012, 22, 65–71. [Google Scholar] [CrossRef]
- Koch, F.; Koch, G. The Molecular Biology of Poliovirus; Springer: Wien, Austria, 1985; p. XVI. [Google Scholar] [CrossRef]
- Mapoles, J.E.; Anderegg, J.W.; Rueckert, R.R. Properties of poliovirus propagated in medium containing cesium chloride: Implications for picornaviral structure. Virology 1978, 90, 103–111. [Google Scholar] [CrossRef]
- Butterworth, B.E.; Grunert, R.R.; Korant, B.D.; Lonberg-Holm, K.; Yin, F.H. Replication of rhinoviruses. Arch. Virol. 1976, 51, 169–189. [Google Scholar] [CrossRef]
- Mestre-Fos, S.; Penev, P.I.; Suttapitugsakul, S.; Hu, M.; Ito, C.; Petrov, A.S.; Wartell, R.M.; Wu, R.; Williams, L.D. G-Quadruplexes in Human Ribosomal RNA. J. Mol. Biol. 2019, 431, 1940–1955. [Google Scholar] [CrossRef]
- Fout, G.S.; Medappa, K.C.; Mapoles, J.E.; Rueckert, R.R. Radiochemical determination of polyamines in poliovirus and human rhinovirus 14. J. Biol. Chem. 1984, 259, 3639–3643. [Google Scholar] [CrossRef]
- Li, P.; Wei, Y.; Mei, M.; Tang, L.; Sun, L.; Huang, W.; Zhou, J.; Zou, C.; Zhang, S.; Qin, C.-F.; et al. Integrative Analysis of Zika Virus Genome RNA Structure Reveals Critical Determinants of Viral Infectivity. Cell Host Microbe 2018, 24, 875–886.e5. [Google Scholar] [CrossRef] [PubMed]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e5. [Google Scholar] [CrossRef] [PubMed]
- Silvers, R.; Keller, H.; Schwalbe, H.; Hengesbach, M. Differential scanning fluorimetry for monitoring RNA stability. ChemBioChem 2015, 16, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Gudnason, H.; Dufva, M.; Bang, D.D.; Wolff, A. Comparison of multiple DNA dyes for real-time PCR: Effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007, 35, e127. [Google Scholar] [CrossRef]
- Khristova, M.L.; Sokolova, M.V.; Yaroslavtseva, N.G.; Busse, T.L.; Kharitonenkov, I.G. Secondary structures of influenza and Sendai virus RNAs. Arch. Virol. 1980, 66, 241–253. [Google Scholar] [CrossRef]
- Ghribi, S.; Maurel, M.-C.; Rougee, M.; Favre, A. Evidence for tertiary structure in natural single stranded RNAs in solution. Nucleic Acids Res. 1988, 16, 1095–1112. [Google Scholar] [CrossRef]
- Kwok, C.K.; Marsico, G.; Sahakyan, A.B.; Chambers, V.S.; Balasubramanian, S. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 2016, 13, 841–844. [Google Scholar] [CrossRef]
- Zou, M.; Li, J.-Y.; Zhang, M.-J.; Li, J.-H.; Huang, J.-T.; You, P.-D.; Liu, S.-W.; Zhou, C.-Q. G-quadruplex binder pyridostatin as an effective multi-target ZIKV inhibitor. Int. J. Biol. Macromol. 2021, 190, 178–188. [Google Scholar] [CrossRef]
- Brabec-Zaruba, M.; Pfanzagl, B.; Blaas, D.; Fuchs, R. Site of Human Rhinovirus RNA Uncoating Revealed by Fluorescent In Situ Hybridization. J. Virol. 2009, 83, 3770–3777. [Google Scholar] [CrossRef]
- Krenn, B.M.; Holzer, B.; Gaudernak, E.; Triendl, A.; van Kuppeveld, F.J.; Seipelt, J. Inhibition of Polyprotein Processing and RNA Replication of Human Rhinovirus by Pyrrolidine Dithiocarbamate Involves Metal Ions. J. Virol. 2005, 79, 13892–13899. [Google Scholar] [CrossRef]
- Real-Hohn, A.; Provance, D.W., Jr.; Gonçalves, R.B.; Denani, C.B.; de Oliveira, A.C.; Salerno, V.P.; Oliveira Gomes, A.M. Impairing the function of MLCK, myosin Va or myosin Vb disrupts Rhinovirus B14 replication. Sci. Rep. 2017, 7, 17153. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Gruenberg, J. Ion flux and the function of endosomes and lysosomes: pH is just the start: The flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 2011, 33, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Blaas, D. Uncoating of human rhinoviruses. Rev. Med. Virol. 2010, 20, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Malgowska, M.; Czajczynska, K.; Gudanis, D.; Tworak, A.; Gdaniec, Z. Overview of the RNA G-quadruplex structures. Acta Biochim. Pol. 2017, 63, 609–621. [Google Scholar] [CrossRef]
- Belmonte-Reche, E.; Serrano-Chacon, I.; Gonzalez, C.; Gallo, J.; Banobre-Lopez, M. Potential G-quadruplexes and i-Motifs in the SARS-CoV-2. PLoS ONE 2021, 16, e0250654. [Google Scholar] [CrossRef] [PubMed]
- Deiana, M.; Jamroskovic, J.; Obi, I.; Sabouri, N. Unravelling the cellular emission fingerprint of the benchmark G-quadruplex-interactive compound Phen-DC3. Chem. Commun. 2020, 56, 14251–14254. [Google Scholar] [CrossRef]
- Broo, K.; Wei, J.; Marshall, D.; Brown, F.; Smith, T.J.; Johnson, J.E.; Schneemann, A.; Siuzdak, G. Viral capsid mobility: A dynamic conduit for inactivation. Proc. Natl. Acad. Sci. USA 2001, 98, 2274–2277. [Google Scholar] [CrossRef]
- Speelman, B.; Brooks, B.R.; Post, C.B. Molecular Dynamics Simulations of Human Rhinovirus and an Antiviral Compound. Biophys. J. 2001, 80, 121–129. [Google Scholar] [CrossRef]
- Chandler-Bostock, R.; Mata, C.P.; Bingham, R.J.; Dykeman, E.C.; Meng, B.; Tuthill, T.J.; Rowlands, D.J.; Ranson, N.A.; Twarock, R.; Stockley, P.G. Assembly of infectious enteroviruses depends on multiple, conserved genomic RNA-coat protein contacts. PLoS Pathog. 2020, 16, e1009146. [Google Scholar] [CrossRef]
- Koirala, D.; Dhakal, S.; Ashbridge, B.; Sannohe, Y.; Rodriguez, R.; Sugiyama, H.; Balasubramanian, S.; Mao, H. A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands. Nat. Chem. 2011, 3, 782–787. [Google Scholar] [CrossRef]
- Endoh, T.; Rode, A.B.; Takahashi, S.; Kataoka, Y.; Kuwahara, M.; Sugimoto, N. Real-Time Monitoring of G-Quadruplex Formation during Transcription. Anal. Chem. 2016, 88, 1984–1989. [Google Scholar] [CrossRef]
- Endoh, T.; Sugimoto, N. Conformational Dynamics of the RNA G-Quadruplex and its Effect on Translation Efficiency. Molecules 2019, 24, 1613. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-H.; Ishii, K.; Tahara, T. Microsecond Equilibrium Dynamics of Hairpin-Forming Oligonucleotides Quantified by Two-Color Two-Dimensional Fluorescence Lifetime Correlation Spectroscopy. J. Phys. Chem. B 2020, 124, 10673–10681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.Q.; Balasubramanian, S. The Kinetics and Folding Pathways of Intramolecular G-Quadruplex Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 19297–19308. [Google Scholar] [CrossRef]
- Nugent, C.I.; Johnson, K.L.; Sarnow, P.; Kirkegaard, K. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 1999, 73, 427–435. [Google Scholar] [CrossRef]
- Baird, N.J.; Inglese, J.; Ferré-D’amaré, A.R. Rapid RNA–ligand interaction analysis through high-information content conformational and stability landscapes. Nat. Commun. 2015, 6, 8898. [Google Scholar] [CrossRef]
- Carrillo-Tripp, M.; Saint-Martin, H.; Ortega-Blake, I. A comparative study of the hydration of Na+ and K+ with refined polarizable model potentials. J. Chem. Phys. 2003, 118, 7062–7073. [Google Scholar] [CrossRef]
- Auffinger, P.; D’ascenzo, L.; Ennifar, E. Sodium and Potassium Interactions with Nucleic Acids. Met. Ions Life Sci. 2016, 16, 167–201. [Google Scholar] [CrossRef] [PubMed]
- Rozov, A.; Khusainov, I.; El Omari, K.; Duman, R.; Mykhaylyk, V.; Yusupov, M.; Westhof, E.; Wagner, A.; Yusupova, G. Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat. Commun. 2019, 10, 2519. [Google Scholar] [CrossRef]
- Uchida, T.; He, Q.; Ralston, C.Y.; Brenowitz, M.; Chance, M.R. Linkage of Monovalent and Divalent Ion Binding in the Folding of the P4−P6 Domain of the Tetrahymena Ribozyme. Biochemistry 2002, 41, 5799–5806. [Google Scholar] [CrossRef]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.; Rowlands, D.J.; Stonehouse, N.; Hogle, J.; Tuthill, T.J. Capsid Protein VP4 of Human Rhinovirus Induces Membrane Permeability by the Formation of a Size-Selective Multimeric Pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef]
- Hillen, H.S.; Parshin, A.V.; Agaronyan, K.; Morozov, Y.I.; Graber, J.J.; Chernev, A.; Schwinghammer, K.; Urlaub, H.; Anikin, M.; Cramer, P.; et al. Mechanism of Transcription Anti-termination in Human Mitochondria. Cell 2017, 171, 1082–1093.e5. [Google Scholar] [CrossRef] [PubMed]
- Indelicato, G.; Cermelli, P.; Twarock, R. A coarse-grained model of the expansion of the human rhinovirus 2 capsid reveals insights in genome release. J. R. Soc. Interface 2019, 16, 20190044. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.C.; Chen, S.B.; Dai, J.; Yuan, J.H.; Ou, T.M.; Huang, Z.S.; Tan, J.H. Tracking the Dynamic Folding and Unfolding of RNA G-Quadruplexes in Live Cells. Angew. Chem. Int. Ed. Engl. 2018, 57, 4702–4706. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, O.; Bourdoncle, A.; Boulé, J.-B.; Brosh, R.M., Jr.; Mergny, J.-L. G-quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Wang, P.; Qin, C.-F.; Hu, Y.; Zhou, X.; Wang, G.-C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; et al. Human Enterovirus Nonstructural Protein 2CATPase Functions as Both an RNA Helicase and ATP-Independent RNA Chaperone. PLoS Pathog. 2015, 11, e1005067. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ribo-Oligonucleotide/(G-Score) | Sequence |
|---|---|
| Negative control/(0) | 5′-UUA CCC UUA CCC UUA CCC UUA CCC UUA-3′ |
| miniTERRA (42) | 5′-UUA GGG UUA GGG UUA GGG UUA GGG UUA-3′ |
| G11; base # 2048–2074 (11) | 5′-GGC ACU CAU GUU AUA UGG GAU GUG GGG-3′ |
| G20; base # 1038–1064 (20) | 5′-CCU CAA AGG GUU GGU GGU GGA AAC UAC-3′ |
| Ribo-Oligonucleotides | IC50 (µM) | |
|---|---|---|
| Na+ | K+ | |
| mini TERRA | 19.29 ± 0.25 | 17.23 ± 0.28 |
| G11 (position 2048–2074) | 5.20 ± 0.18 | 9.11 ± 0.29 |
| G20 (position 1038–1064) | 17.13 ± 0.45 | 19.38 ± 0.32 |
| Treatment | PDS Bound (Moles/Mole Virus) | Relative to Maximum |
|---|---|---|
| 34 °C/Na+ | 10.0 | 100% |
| 4 °C/Na+ | 0.9 | 8.8% |
| 34 °C/K+ | 0.2 | 1.4% |
| 4 °C/K+ | 0.4 | 3.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Real-Hohn, A.; Groznica, M.; Kontaxis, G.; Zhu, R.; Chaves, O.A.; Vazquez, L.; Hinterdorfer, P.; Kowalski, H.; Blaas, D. Stabilization of the Quadruplex-Forming G-Rich Sequences in the Rhinovirus Genome Inhibits Uncoating—Role of Na+ and K+. Viruses 2023, 15, 1003. https://doi.org/10.3390/v15041003
Real-Hohn A, Groznica M, Kontaxis G, Zhu R, Chaves OA, Vazquez L, Hinterdorfer P, Kowalski H, Blaas D. Stabilization of the Quadruplex-Forming G-Rich Sequences in the Rhinovirus Genome Inhibits Uncoating—Role of Na+ and K+. Viruses. 2023; 15(4):1003. https://doi.org/10.3390/v15041003
Chicago/Turabian StyleReal-Hohn, Antonio, Martin Groznica, Georg Kontaxis, Rong Zhu, Otávio Augusto Chaves, Leonardo Vazquez, Peter Hinterdorfer, Heinrich Kowalski, and Dieter Blaas. 2023. "Stabilization of the Quadruplex-Forming G-Rich Sequences in the Rhinovirus Genome Inhibits Uncoating—Role of Na+ and K+" Viruses 15, no. 4: 1003. https://doi.org/10.3390/v15041003
APA StyleReal-Hohn, A., Groznica, M., Kontaxis, G., Zhu, R., Chaves, O. A., Vazquez, L., Hinterdorfer, P., Kowalski, H., & Blaas, D. (2023). Stabilization of the Quadruplex-Forming G-Rich Sequences in the Rhinovirus Genome Inhibits Uncoating—Role of Na+ and K+. Viruses, 15(4), 1003. https://doi.org/10.3390/v15041003