

Surveillance, Isolation, and Genetic Characterization of Bat Herpesviruses in Zambia
 , ,
, ,  , , , , , ,
, , , , , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Amplification of the Herpesvirus DNA Polymerase Gene by PCR
2.3. Virus Isolation
2.4. Genome Sequencing, Construction, and Annotation
2.5. Genetic Comparison and Phylogenetic Analysis
3. Results
3.1. Screening for Herpesviruses with DPOL Gene by Semi-Nested PCR
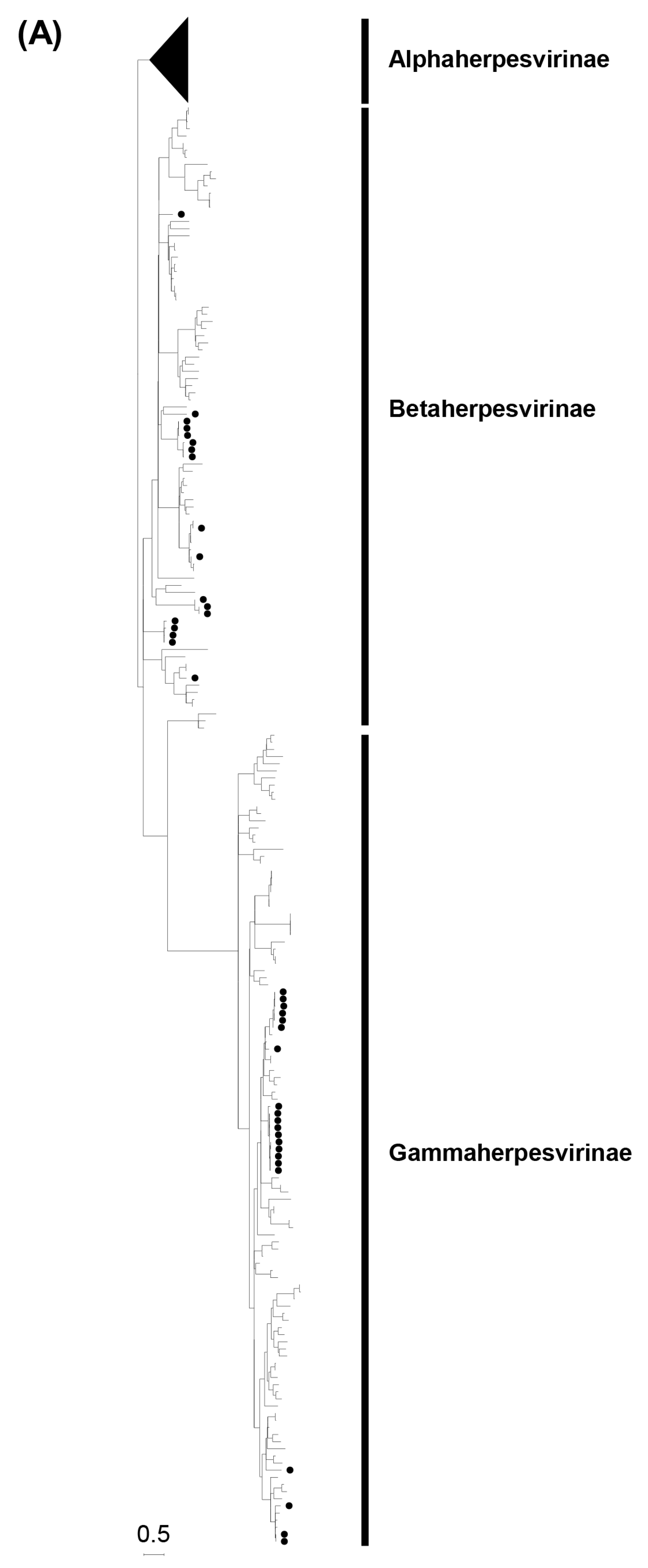
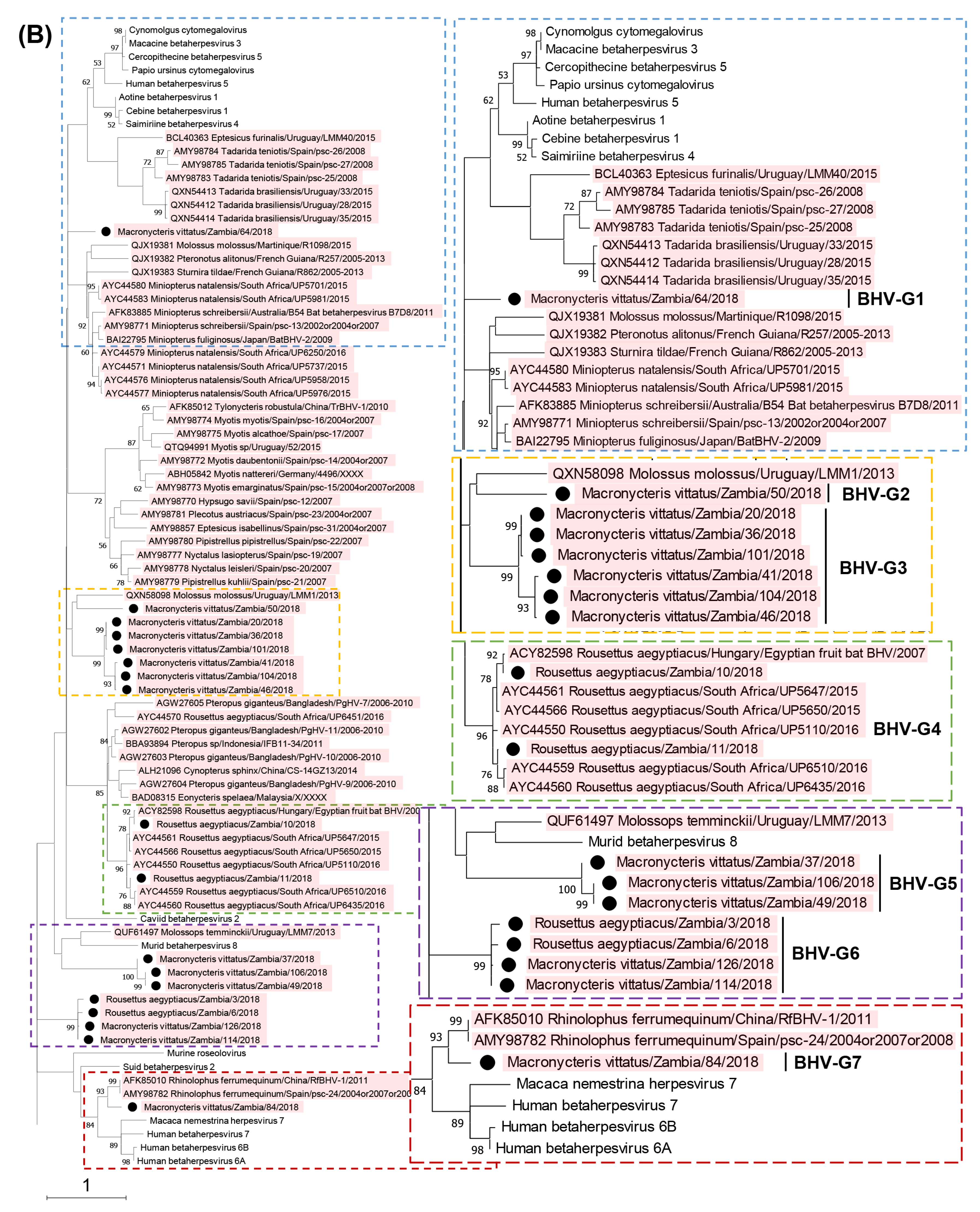
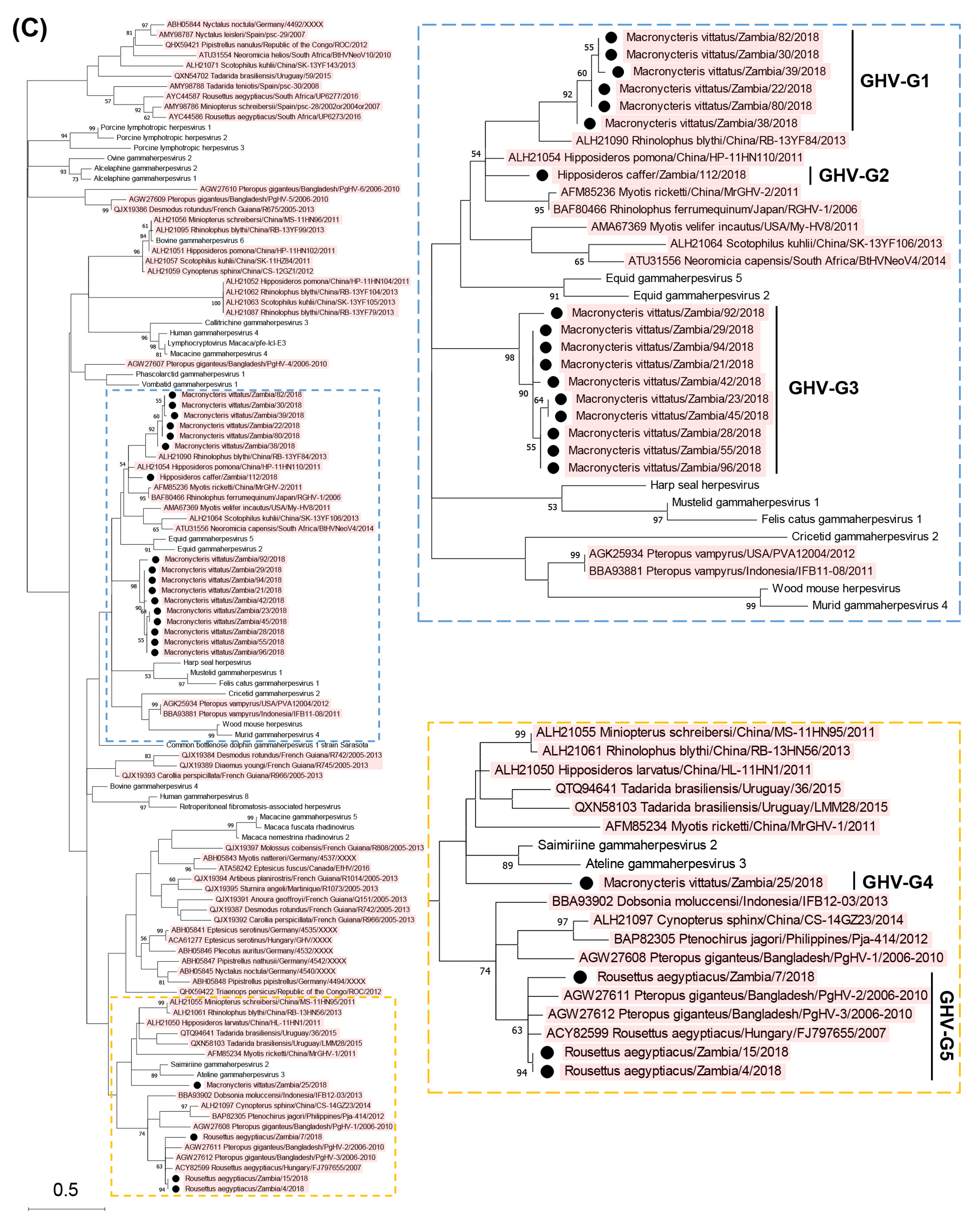
3.2. Phylogenetic Analyses Based on the Partial DPOL Gene
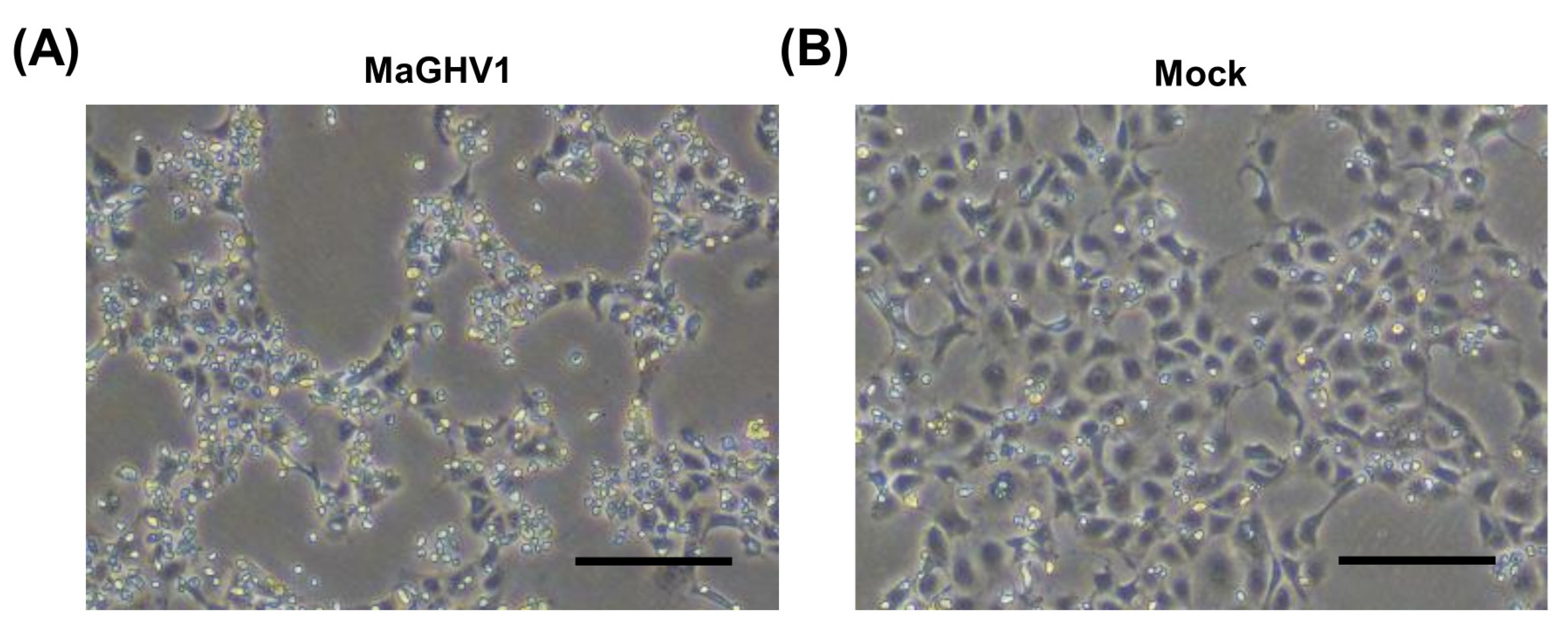
3.3. Isolation of a Novel Gammaherpesvirus from Macronycteris Vittatus Bats
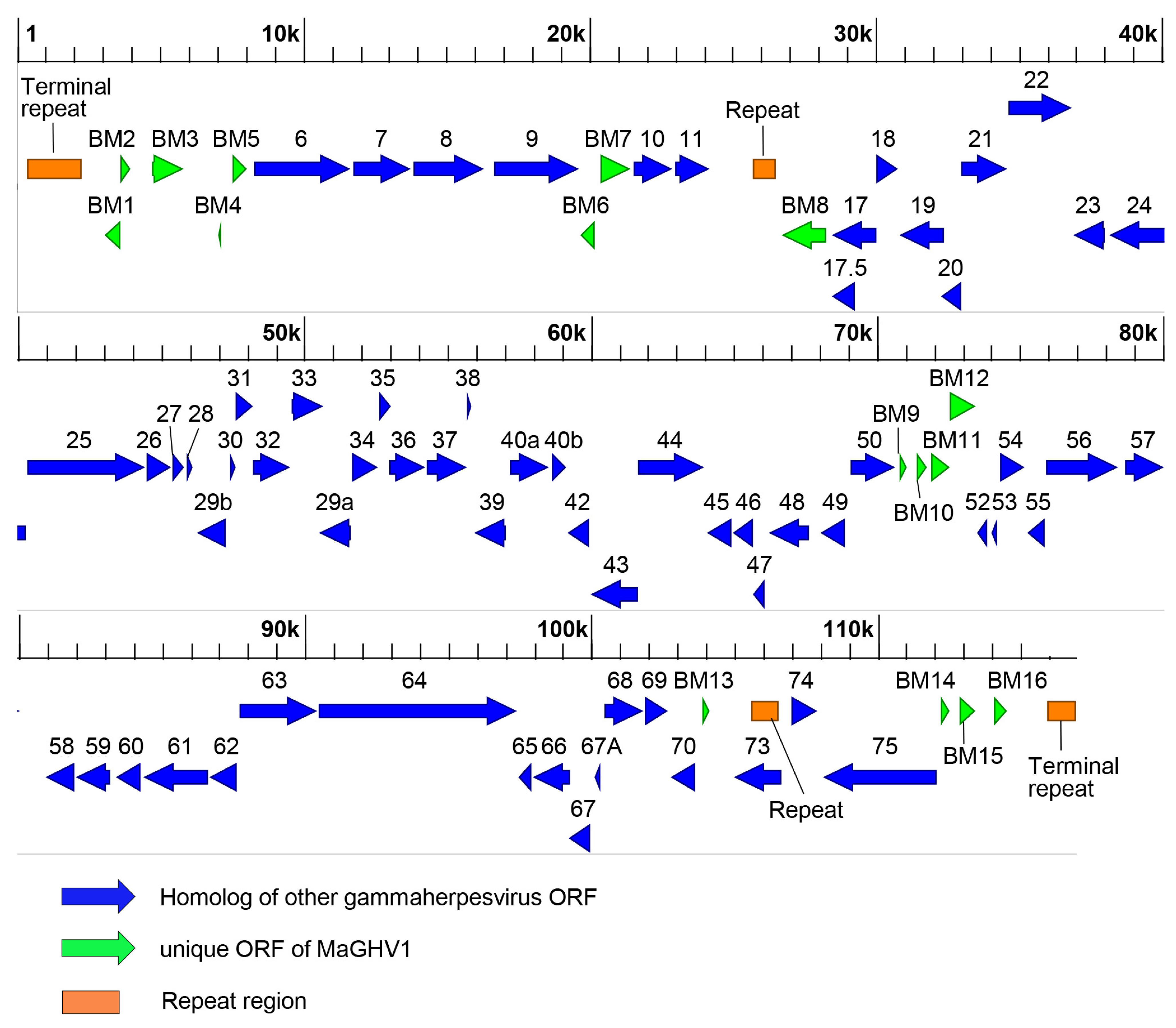
3.4. Complete Genome of MaGHV1 and Genetic Comparison between Strains
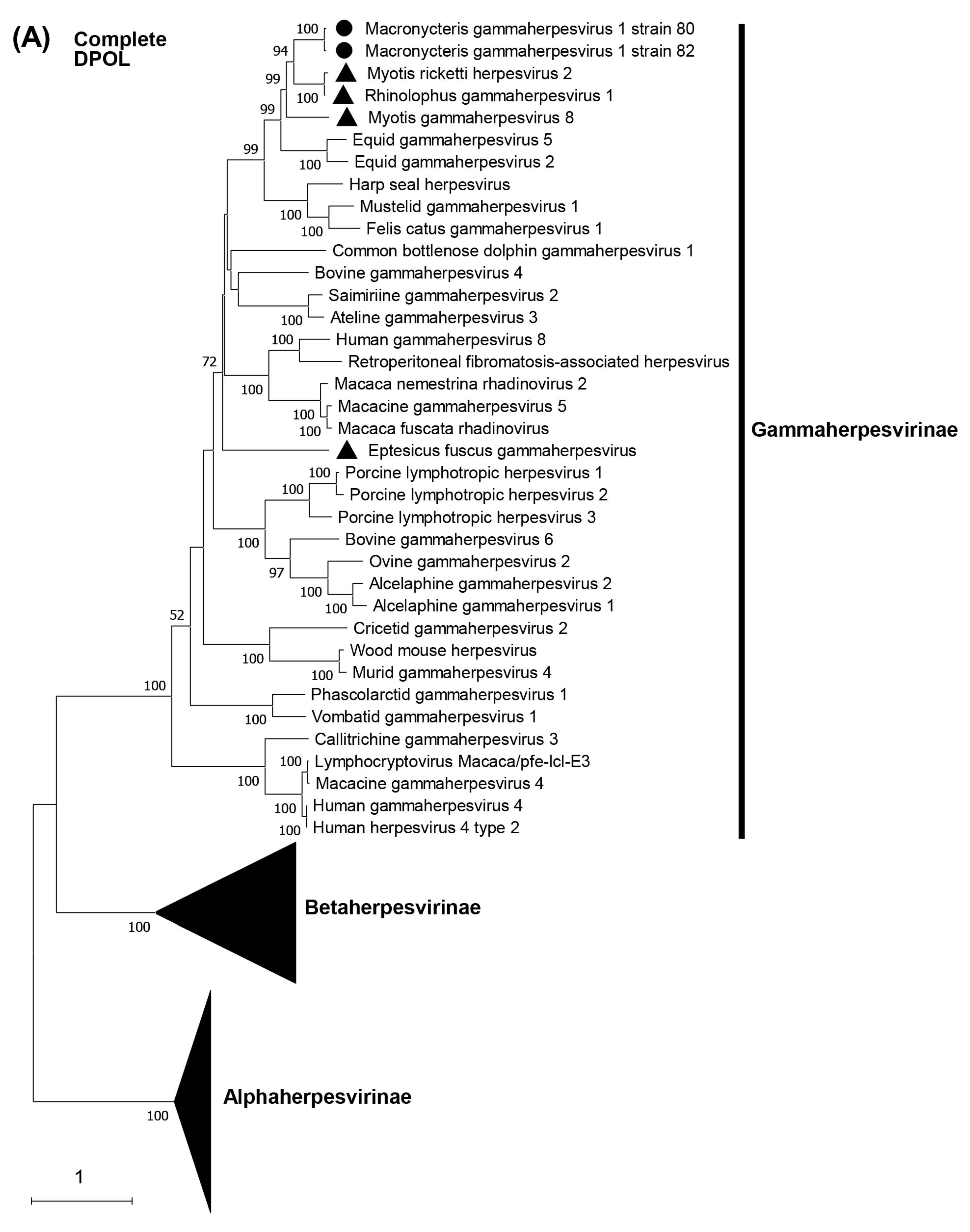
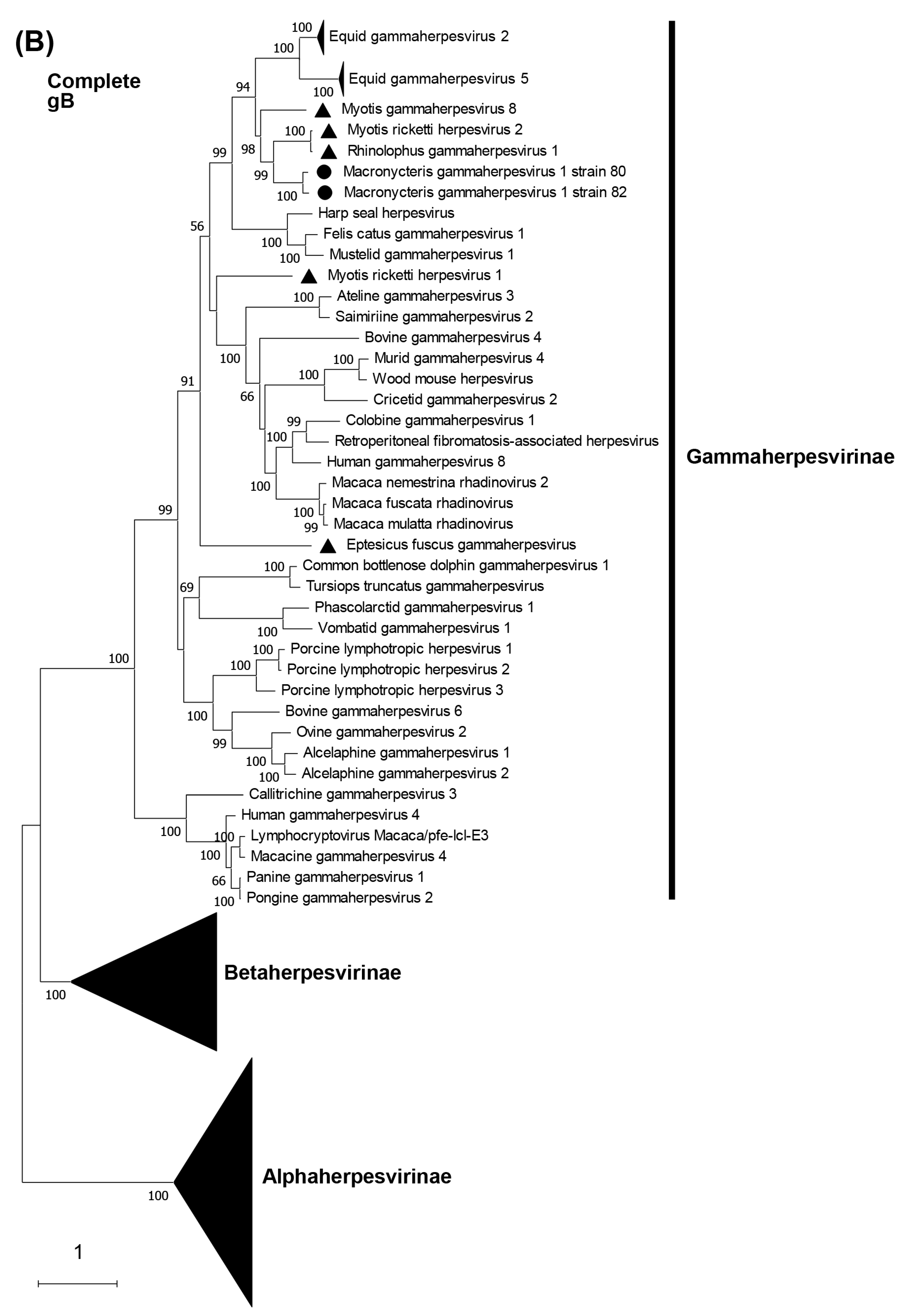
3.5. Phylogenetic Characterization of MaGHV1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, B.; Han, Y.; Wang, Y.; Chen, L.; Wu, Z.; Yang, J. ZOVER: The database of zoonotic and vector-borne viruses. Nucleic Acids Res. 2022, 50, D943–D949. [Google Scholar] [CrossRef] [PubMed]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer/Lippincott Williams & Winlkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1802–1822. [Google Scholar]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef]
- Fossum, E.; Friedel, C.C.; Rajagopala, S.V.; Titz, B.; Baiker, A.; Schmidt, T.; Kraus, T.; Stellberger, T.; Rutenberg, C.; Suthram, S.; et al. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 2009, 5, e1000570. [Google Scholar] [CrossRef] [Green Version]
- James, S.; Donato, D.; de Thoisy, B.; Lavergne, A.; Lacoste, V. Novel herpesviruses in neotropical bats and their relationship with other members of the Herpesviridae family. Infect. Genet. Evol. 2020, 84, 104367. [Google Scholar] [CrossRef]
- Cameron, K.; Hayes, B.; Olson, S.H.; Smith, B.R.; Pante, J.; Laudisoit, A.; Goldstein, T.; Joly, D.O.; Bagamboula MPassi, R.; Lange, C.E. Detection of first gammaherpesvirus sequences in Central African bats. N. Microbes N. Infect. 2020, 36, 100705. [Google Scholar] [CrossRef]
- Kamau, J.; Ergunay, K.; Webala, P.W.; Justi, S.A.; Bourke, B.P.; Kamau, M.W.; Hassell, J.; Chege, M.N.; Mwaura, D.K.; Simiyu, C.; et al. A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats. Viruses 2022, 14, 2820. [Google Scholar] [CrossRef]
- Dietrich, M.; Kearney, T.; Seamark, E.C.J.; Paweska, J.T.; Markotter, W. Synchronized shift of oral, faecal and urinary microbiotas in bats and natural infection dynamics during seasonal reproduction. R. Soc. Open Sci. 2018, 5, 180041. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-coloured fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Razafindratsimandresy, R.; Jeanmaire, E.M.; Counor, D.; Vasconcelos, P.F.; Sall, A.A.; Reynes, J.M. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J. Gen. Virol. 2009, 90, 44–47. [Google Scholar] [CrossRef]
- Sasaki, M.; Setiyono, A.; Handharyani, E.; Kobayashi, S.; Rahmadani, I.; Taha, S.; Adiani, S.; Subangkit, M.; Nakamura, I.; Sawa, H.; et al. Isolation and characterization of a novel alphaherpesvirus in fruit bats. J. Virol. 2014, 88, 9819–9829. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Yamada, S.; Fujii, H.; Yoshikawa, T.; Shibamura, M.; Harada, S.; Fukushi, S.; Le, M.Q.; Nguyen, C.T.; Nguyen, T.T.T.; et al. Characterization of a Novel Alphaherpesvirus Isolated from the Fruit Bat. J. Virol. 2020, 94, e00673-20. [Google Scholar] [CrossRef]
- Watanabe, S.; Maeda, K.; Suzuki, K.; Ueda, N.; Iha, K.; Taniguchi, S.; Shimoda, H.; Kato, K.; Yoshikawa, Y.; Morikawa, S.; et al. Novel betaherpesvirus in bats. Emerg. Infect. Dis. 2010, 16, 986–988. [Google Scholar] [CrossRef]
- Zhang, H.; Todd, S.; Tachedjian, M.; Barr, J.A.; Luo, M.; Yu, M.; Marsh, G.A.; Crameri, G.; Wang, L.F. A novel bat herpesvirus encodes homologues of major histocompatibility complex classes I and II, C-type lectin, and a unique family of immune-related genes. J. Virol. 2012, 86, 8014–8030. [Google Scholar] [CrossRef] [Green Version]
- Shabman, R.S.; Shrivastava, S.; Tsibane, T.; Attie, O.; Jayaprakash, A.; Mire, C.E.; Dilley, K.E.; Puri, V.; Stockwell, T.B.; Geisbert, T.W.; et al. Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line. mSphere 2016, 1, e00070-15. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, S.; Rapin, N.; Dorville, N.; Hill, J.E.; Town, J.; Willis, C.K.R.; Bollinger, T.K.; Misra, V. Isolation, characterization and prevalence of a novel Gammaherpesvirus in Eptesicus fuscus, the North American big brown bat. Virology 2018, 516, 227–238. [Google Scholar] [CrossRef]
- Noguchi, K.; Kuwata, R.; Shimoda, H.; Mizutani, T.; Hondo, E.; Maeda, K. The complete genomic sequence of Rhinolophus gammaherpesvirus 1 isolated from a greater horseshoe bat. Arch. Virol. 2019, 164, 317–319. [Google Scholar] [CrossRef]
- Kajihara, M.; Hang’ombe, B.M.; Changula, K.; Harima, H.; Isono, M.; Okuya, K.; Yoshida, R.; Mori-Kajihara, A.; Eto, Y.; Orba, Y.; et al. Marburgvirus in Egyptian Fruit Bats, Zambia. Emerg. Infect. Dis. 2019, 25, 1577–1580. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Nakao, R.; Hang’ombe, B.M.; Sato, K.; Kajihara, M.; Kanchela, S.; Changula, K.; Eto, Y.; Ndebe, J.; Sasaki, M.; et al. Human Borreliosis Caused by a New World Relapsing Fever Borrelia-like Organism in the Old World. Clin. Infect. Dis. 2019, 69, 107–112. [Google Scholar] [CrossRef]
- Rose, T.M.; Strand, K.B.; Schultz, E.R.; Schaefer, G.; Rankin, G.W.; Thouless, M.E.; Tsai, C.C.; Bosch, M.L. Identification of two homologs of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in retroperitoneal fibromatosis of different macaque species. J. Virol. 1997, 71, 4138–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Telford, E.A.; Watson, M.S.; Aird, H.C.; Perry, J.; Davison, A.J. The DNA sequence of equine herpesvirus 2. J. Mol. Biol. 1995, 249, 520–528. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [Green Version]
- Wada, Y.; Sasaki, M.; Setiyono, A.; Handharyani, E.; Rahmadani, I.; Taha, S.; Adiani, S.; Latief, M.; Kholilullah, Z.A.; Subangkit, M.; et al. Detection of novel gammaherpesviruses from fruit bats in Indonesia. J. Med. Microbiol. 2018, 67, 415–422. [Google Scholar] [CrossRef]
- Moreira Marrero, L.; Botto Nuñez, G.; Malta, L.; Delfraro, A.; Frabasile, S. Ecological and Conservation Significance of Herpesvirus Infection in Neotropical Bats. Ecohealth 2021, 18, 123–133. [Google Scholar] [CrossRef]
- Azab, W.; Dayaram, A.; Greenwood, A.D.; Osterrieder, N. How Host Specific Are Herpesviruses? Lessons from Herpesviruses Infecting Wild and Endangered Mammals. Annu. Rev. Virol. 2018, 5, 53–68. [Google Scholar] [CrossRef]
- Griffiths, M.E.; Bergner, L.M.; Broos, A.; Meza, D.K.; Filipe, A.D.S.; Davison, A.; Tello, C.; Becker, D.J.; Streicker, D.G. Epidemiology and biology of a herpesvirus in rabies endemic vampire bat populations. Nat. Commun. 2020, 11, 5951. [Google Scholar] [CrossRef]
- Escalera-Zamudio, M.; Rojas-Anaya, E.; Kolokotronis, S.O.; Taboada, B.; Loza-Rubio, E.; Méndez-Ojeda, M.L.; Arias, C.F.; Osterrieder, N.; Greenwood, A.D. Bats, Primates, and the Evolutionary Origins and Diversification of Mammalian Gammaherpesviruses. mBio 2016, 7, e01425-16. [Google Scholar] [CrossRef] [Green Version]
- ACR. African Chiroptera Report 2022; Cakenberghe, V.V., Seamark, E.C.J., Eds.; AfricanBats NPC: Pretoria, South Africa, 2022; pp. i–xvii + 8661. [Google Scholar]
- Wright, G.S. Hipposideros caffer (chiroptera: Hipposideridae). Mamm. Species 2009, 845, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [Green Version]
- Hutson, A.M.; Mickleburgh, S.P.; Racey, P.A. Microchiropteran Bats: Global Status Survey and Conservation Action Plan; IUCN: Gland, Switzerland, 2001; Volume 56. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Date | Bat (Species) | Sample ID | No. of Positive/No. of Tested Samples (%) |
|---|---|---|---|
| 19 November | Egyptian fruit bat (Rousettus aegyptiacus) | 1–17 | 7/17 (41.2) |
| Striped leaf-nosed bat (Macronycteris vittatus) | 18–70 | 39/53 (73.6) | |
| 22 November | Egyptian fruit bat (Rousettus aegyptiacus) | 71–77 | 0/7 (0) |
| Striped leaf-nosed bat (Macronycteris vittatus) | 78–111 113–130 | 43/52 (82.7) | |
| Sundevall’s roundleaf bat (Hipposideros caffer) | 112 | 1/1 (100) | |
| Total | 90/130 (69.2) | ||
| Virus | Accession No. | Sample ID | Subfamily | Group |
|---|---|---|---|---|
| Macronycteris vittatus/Zambia/64/2018 | LC762209 | 64 | Betaherpesvirinae | BHV-G1 |
| Macronycteris vittatus/Zambia/50/2018 | LC762210 | 50, 69 | Betaherpesvirinae | BHV-G2 |
| Macronycteris vittatus/Zambia/20/2018 | LC762211 | 20 | Betaherpesvirinae | BHV-G3 |
| Macronycteris vittatus/Zambia/36/2018 | LC762212 | 36 | Betaherpesvirinae | BHV-G3 |
| Macronycteris vittatus/Zambia/41/2018 | LC762213 | 41 | Betaherpesvirinae | BHV-G3 |
| Macronycteris vittatus/Zambia/46/2018 | LC762214 | 46 | Betaherpesvirinae | BHV-G3 |
| Macronycteris vittatus/Zambia/101/2018 | LC762215 | 101, 111 | Betaherpesvirinae | BHV-G3 |
| Macronycteris vittatus/Zambia/104/2018 | LC762216 | 104 | Betaherpesvirinae | BHV-G3 |
| Rousettus aegyptiacus/Zambia/10/2018 | LC762217 | 10 | Betaherpesvirinae | BHV-G4 |
| Rousettus aegyptiacus/Zambia/11/2018 | LC762218 | 11 | Betaherpesvirinae | BHV-G4 |
| Macronycteris vittatus/Zambia/37/2018 | LC762219 | 37, 44 | Betaherpesvirinae | BHV-G5 |
| Macronycteris vittatus/Zambia/49/2018 | LC762220 | 49, 56, 61, 66, 89, 103, 121, 123, 128 | Betaherpesvirinae | BHV-G5 |
| Macronycteris vittatus/Zambia/106/2018 | LC762221 | 106, 109 | Betaherpesvirinae | BHV-G5 |
| Rousettus aegyptiacus/Zambia/3/2018 | LC762222 | 3 | Betaherpesvirinae | BHV-G6 |
| Rousettus aegyptiacus/Zambia/6/2018 | LC762223 | 6 | Betaherpesvirinae | BHV-G6 |
| Macronycteris vittatus/Zambia/114/2018 | LC762224 | 114 | Betaherpesvirinae | BHV-G6 |
| Macronycteris vittatus/Zambia/126/2018 | LC762225 | 126 | Betaherpesvirinae | BHV-G6 |
| Macronycteris vittatus/Zambia/84/2018 | LC762226 | 84 | Betaherpesvirinae | BHV-G7 |
| Macronycteris vittatus/Zambia/22/2018 | LC762227 | 22, 58, 65, 81, 83, 110, 130 | Gammaherpesvirinae | GHV-G1 |
| Macronycteris vittatus/Zambia/30/2018 | LC762228 | 30, 60 | Gammaherpesvirinae | GHV-G1 |
| Macronycteris vittatus/Zambia/38/2018 | LC762229 | 38 | Gammaherpesvirinae | GHV-G1 |
| Macronycteris vittatus/Zambia/39/2018 | LC762230 | 39 | Gammaherpesvirinae | GHV-G1 |
| Macronycteris vittatus/Zambia/80/2018 | LC762231 | 59, 70, 80, 86, 91, 97, 98, 107, 113 | Gammaherpesvirinae | GHV-G1 |
| Macronycteris vittatus/Zambia/82/2018 | LC762232 | 18, 24, 26, 31, 78, 82, 85, 88, 124, 129 | Gammaherpesvirinae | GHV-G1 |
| Hipposideros caffer/Zambia/112/2018 | LC762233 | 112 | Gammaherpesvirinae | GHV-G2 |
| Macronycteris vittatus/Zambia/21/2018 | LC762234 | 21 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/23/2018 | LC762235 | 23, 43, 118 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/28/2018 | LC762236 | 28, 99, 100, 116, 120 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/29/2018 | LC762237 | 29 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/42/2018 | LC762238 | 42 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/45/2018 | LC762239 | 45 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/55/2018 | LC762240 | 55, 119 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/92/2018 | LC762241 | 92 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/94/2018 | LC762242 | 94 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/96/2018 | LC762243 | 96 | Gammaherpesvirinae | GHV-G3 |
| Macronycteris vittatus/Zambia/25/2018 | LC762244 | 25, 33, 34, 47, 52, 95, 105, 108, 125 | Gammaherpesvirinae | GHV-G4 |
| Rousettus aegyptiacus/Zambia/4/2018 | LC762245 | 4 | Gammaherpesvirinae | GHV-G5 |
| Rousettus aegyptiacus/Zambia/7/2018 | LC762246 | 7 | Gammaherpesvirinae | GHV-G5 |
| Rousettus aegyptiacus/Zambia/15/2018 | LC762247 | 15 | Gammaherpesvirinae | GHV-G5 |
| Gene | Location (Nucleotides) | Strand | Size (No. of Amino Acids) | Product or Predicted Function |
|---|---|---|---|---|
| BM1 | 3031–3630 | − | 199 | homolog of CASP8 and FADD-like apoptosis regulator |
| BM2 | 3577–3990 | + | 137 | hypothetical protein |
| BM3 | 4693–5832 | + | 379 | homolog of EHV2 E3 membrane protein E3 |
| BM4 | 6988–7152 | − | 54 | homolog of E3 ubiquitin-protein ligase and RGHV1 ORF4 MIR-like membrane protein |
| BM5 | 7520–8077 | + | 185 | homolog of EHV2 E4 apoptosis regulator BALF1 |
| ORF6 | 8279–11,677 | + | 1132 | single-stranded DNA binding protein |
| ORF7 | 11,748–13,799 | + | 683 | DNA packaging terminase subunit 2 |
| ORF8 | 13,816–16,362 | + | 848 | glycoprotein B |
| ORF9 | 16,633–19,638 | + | 1001 | DNA polymerase catalytic subunit |
| BM6 | 19,673–20,185 | − | 170 | bcl-2-like protein |
| BM7 | 20,383–21,486 | + | 367 | homolog of EHV2 E6 membrane protein BILF1 |
| ORF10 | 21,530–22,927 | + | 465 | homolog of EHV2 ORF10 protein G10 |
| ORF11 | 22,977–24,209 | + | 410 | homolog of EHV2 ORF11 virion protein G11 |
| BM8 | 26,676–28,304 | − | 542 | homolog of bovine herpesvirus 6 Bov8 putative major envelope glycoprotein, RGHV1 ORF18 glycoprotein |
| ORF17 | 28,449–30,029 | − | 526 | capsid maturation protease |
| ORF17.5 | 28,449–29,300 | − | 284 | capsid scaffold protein |
| ORF18 | 30,022–30,798 | + | 258 | homolog of EHV2 ORF18 protein UL79 |
| ORF19 | 30,795–32,417 | − | 540 | homolog of EHV2 ORF19 DNA packaging tegument protein UL25 |
| ORF20 | 32,281–32,985 | − | 234 | homolog of EHV2 ORF20 nuclear protein UL24 |
| ORF21 | 32,984–34,600 | + | 538 | thymidine kinase |
| ORF22 | 34,600–36,861 | + | 753 | glycoprotein H |
| ORF23 | 36,858–38,051 | − | 397 | homolog of EHV2 ORF23 tegument protein UL88 |
| ORF24 | 38,106–40,298 | − | 730 | homolog of EHV2 ORF24 protein UL87 |
| ORF25 | 40,303–44,436 | + | 1377 | major capsid protein |
| ORF26 | 44,456–45,358 | + | 300 | capsid triplex subunit 2 |
| ORF27 | 45,359–45,802 | + | 147 | homolog of EHV2 ORF27 envelope glycoprotein 48 |
| ORF28 | 45,867–46,118 | + | 83 | homolog of EHV2 ORF28 envelope glycoprotein 150 |
| ORF29a | 50,460–51,623 | − | 387 | DNA packaging terminase subunit 1 |
| ORF29b | 46,207–47,250 | − | 347 | DNA packaging terminase subunit 1 |
| ORF30 | 47,385–47,639 | + | 84 | homolog of EHV2 ORF30 protein UL91 |
| ORF31 | 47,552–48,235 | + | 227 | homolog of EHV2 ORF31 protein UL92 |
| ORF32 | 48,181–49,536 | + | 451 | homolog of EHV2 ORF32 DNA packaging tegument protein UL17 |
| ORF33 | 49,529–50,695 | + | 388 | homolog of EHV2 ORF33 tegument protein UL16 |
| ORF34 | 51,622–52,590 | + | 322 | homolog of EHV2 ORF34 protein UL95 |
| ORF35 | 52,577–53,047 | + | 156 | homolog of EHV2 ORF35 tegument protein UL14 |
| ORF36 | 52,944–54,245 | + | 433 | tegument serine/threonine protein kinase |
| ORF37 | 54,248–55,702 | + | 484 | deoxyribonuclease |
| ORF38 | 55,657–55,854 | + | 65 | myristylated tegument protein |
| ORF39 | 55,918–57,051 | − | 377 | glycoprotein M |
| ORF40a | 57,162–58,538 | + | 458 | helicase-primase subunit |
| ORF40b | 58,624–59,154 | + | 176 | helicase-primase subunit |
| ORF42 | 59,149–59,964 | − | 271 | homolog of EHV2 ORF42 tegument protein UL7 |
| ORF43 | 59,951–61,654 | − | 567 | capsid portal protein |
| ORF44 | 61,620–63,986 | + | 788 | helicase-primase helicase subunit |
| ORF45 | 64,035–64,934 | − | 299 | homolog of EHV2 ORF45 tegument protein G45 |
| ORF46 | 64,936–65,694 | − | 252 | uracil-DNA glycosylase |
| ORF47 | 65,657–66,115 | − | 152 | glycoprotein L |
| ORF48 | 66,213–67,646 | − | 477 | homolog of EHV2 ORF48 tegument protein G48 |
| ORF49 | 68,010–68,894 | − | 294 | homolog of EHV2 ORF49 tegument protein G49 |
| ORF50 | 69,039–70,646 | + | 535 | protein Rta |
| BM9 | 70,780–71,067 | + | 95 | hypothetical protein, homolog of RGHV1 ORF53 |
| BM10 | 71,349–71,759 | + | 136 | hypothetical protein |
| BM11 | 71,849–72,568 | + | 239 | homolog of EHV2 E7A envelope glycoprotein 42 |
| BM12 | 72,526–73,479 | + | 317 | hypothetical protein, homolog of RGHV1 ORF57 |
| ORF52 | 73,493–73,897 | − | 134 | homolog of EHV2 ORF52 virion protein G52 |
| ORF53 | 73,952–74,212 | − | 86 | glycoprotein N |
| ORF54 | 74,302–75,171 | + | 289 | dUTPase |
| ORF55 | 75,240–75,893 | − | 217 | homolog of EHV2 ORF55 tegument protein UL51 |
| ORF56 | 75,866–78,433 | + | 855 | helicase-primase primase subunit |
| ORF57 | 78,642–80,066 | + | 474 | multifunctional expression regulator |
| ORF58 | 80,927–81,964 | − | 345 | homolog of EHV2 ORF58 envelope protein UL43 |
| ORF59 | 81,973–83,202 | − | 409 | DNA polymerase processivity subunit |
| ORF60 | 83,364–84,281 | − | 305 | ribonucleotide reductase subunit 2 |
| ORF61 | 84,309–86,621 | − | 770 | ribonucleotide reductase subunit 1 |
| ORF62 | 86,645–87,646 | − | 333 | capsid triplex subunit 1 |
| ORF63 | 87,657–90,434 | + | 925 | homolog of EHV2 ORF63 tegument protein UL37 |
| ORF64 | 90,450–97,421 | + | 2323 | large tegument protein |
| ORF65 | 97,435–97,935 | − | 166 | small capsid protein |
| ORF66 | 97,946–99,256 | − | 436 | homolog of EHV2 ORF66 protein UL49 |
| ORF67 | 99,163–100,002 | − | 279 | nuclear egress membrane protein |
| ORF67A | 100,065–100,334 | − | 89 | homolog of EHV2 ORF67A DNA packaging protein UL33 |
| ORF68 | 100,437–101,819 | + | 460 | envelope glycoprotein, DNA packaging protein UL32 |
| ORF69 | 101,821–102,684 | + | 287 | nuclear egress lamina protein |
| ORF70 | 102,750–103,628 | − | 292 | thymidylate synthase |
| BM13 | 103,853–104,161 | + | 102 | hypothetical protein |
| ORF73 | 104,958–106,661 | − | 567 | nuclear antigen LANA-1 |
| ORF74 | 106,940–107,914 | + | 324 | homolog of EHV2 ORF74 membrane protein G74 |
| ORF75 | 108,078–112,091 | − | 1337 | homolog of EHV2 ORF75 tegument protein G75 |
| BM14 | 112,188–112,517 | + | 109 | hypothetical protein |
| BM15 | 112,844–113,437 | + | 197 | hypothetical protein |
| BM16 | 114,035–114,538 | + | 167 | hypothetical protein |
| Terminal repeat | 319–2295 | none | none | none |
| Repeat region 1 | 25,665–26,556 | none | none | none |
| Repeat region 2 | 105,538–106,545 | none | none | none |
| Terminal repeat | 115,903–116,972 | none | none | none |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harima, H.; Qiu, Y.; Yamagishi, J.; Kajihara, M.; Changula, K.; Okuya, K.; Isono, M.; Yamaguchi, T.; Ogawa, H.; Nao, N.; et al. Surveillance, Isolation, and Genetic Characterization of Bat Herpesviruses in Zambia. Viruses 2023, 15, 1369. https://doi.org/10.3390/v15061369
Harima H, Qiu Y, Yamagishi J, Kajihara M, Changula K, Okuya K, Isono M, Yamaguchi T, Ogawa H, Nao N, et al. Surveillance, Isolation, and Genetic Characterization of Bat Herpesviruses in Zambia. Viruses. 2023; 15(6):1369. https://doi.org/10.3390/v15061369
Chicago/Turabian StyleHarima, Hayato, Yongjin Qiu, Junya Yamagishi, Masahiro Kajihara, Katendi Changula, Kosuke Okuya, Mao Isono, Tomoyuki Yamaguchi, Hirohito Ogawa, Naganori Nao, and et al. 2023. "Surveillance, Isolation, and Genetic Characterization of Bat Herpesviruses in Zambia" Viruses 15, no. 6: 1369. https://doi.org/10.3390/v15061369
APA StyleHarima, H., Qiu, Y., Yamagishi, J., Kajihara, M., Changula, K., Okuya, K., Isono, M., Yamaguchi, T., Ogawa, H., Nao, N., Sasaki, M., Simulundu, E., Mweene, A. S., Sawa, H., Ishihara, K., Hang’ombe, B. M., & Takada, A. (2023). Surveillance, Isolation, and Genetic Characterization of Bat Herpesviruses in Zambia. Viruses, 15(6), 1369. https://doi.org/10.3390/v15061369