
Ultrastructural and Functional Characterization of Mitochondrial Dynamics Induced by Human Respiratory Syncytial Virus Infection in HEp-2 Cells

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Line Culture, Viral Stock and Treatments
2.2. Transmission Electron Microscopy
2.3. Qualitative Morphological Evaluation
2.4. Morphometric Analysis
2.5. RNA Extraction
2.6. Analysis of Gene Expression by Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.7. hRSV Viral Copy Quantification by Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.8. Statistical Analysis
3. Results
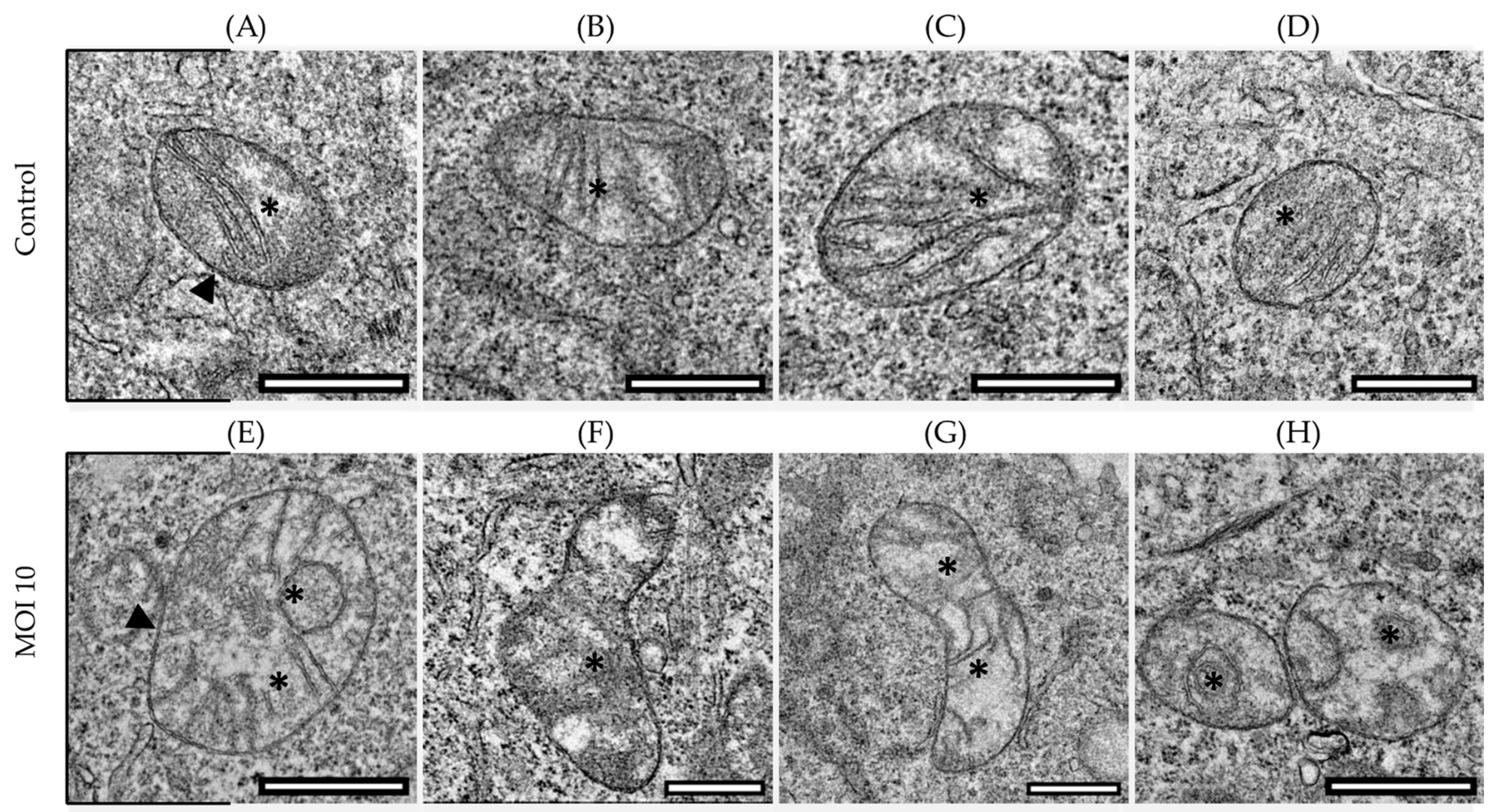
3.1. Effects of hRSV ON1 Genotype Infection on the Morphology of the Mitochondria of the HEp-2 Cell Line
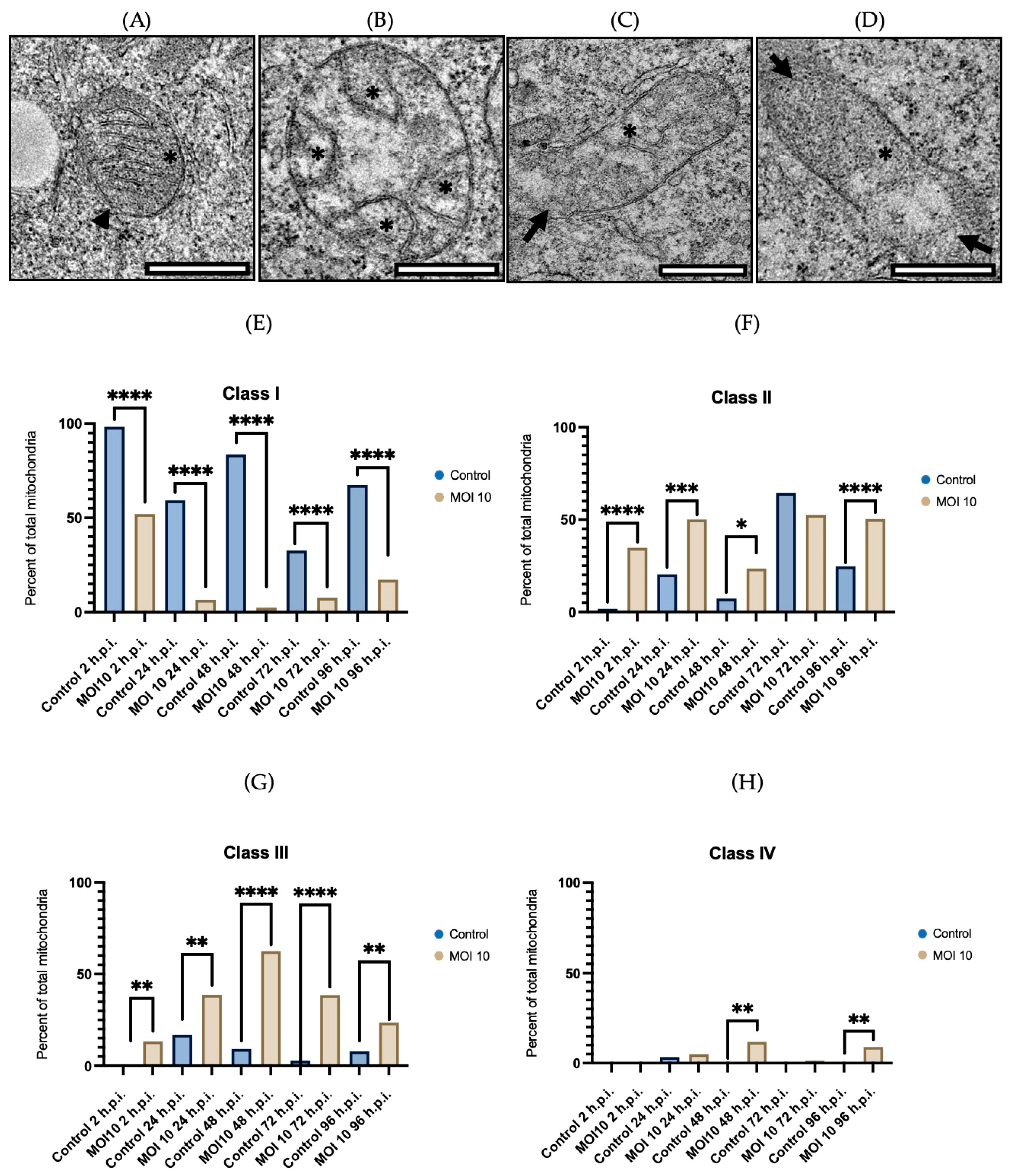
3.2. Morphological Characterization of Mitochondria Infected with RSV ON1 in the HEp-2 Cell Line
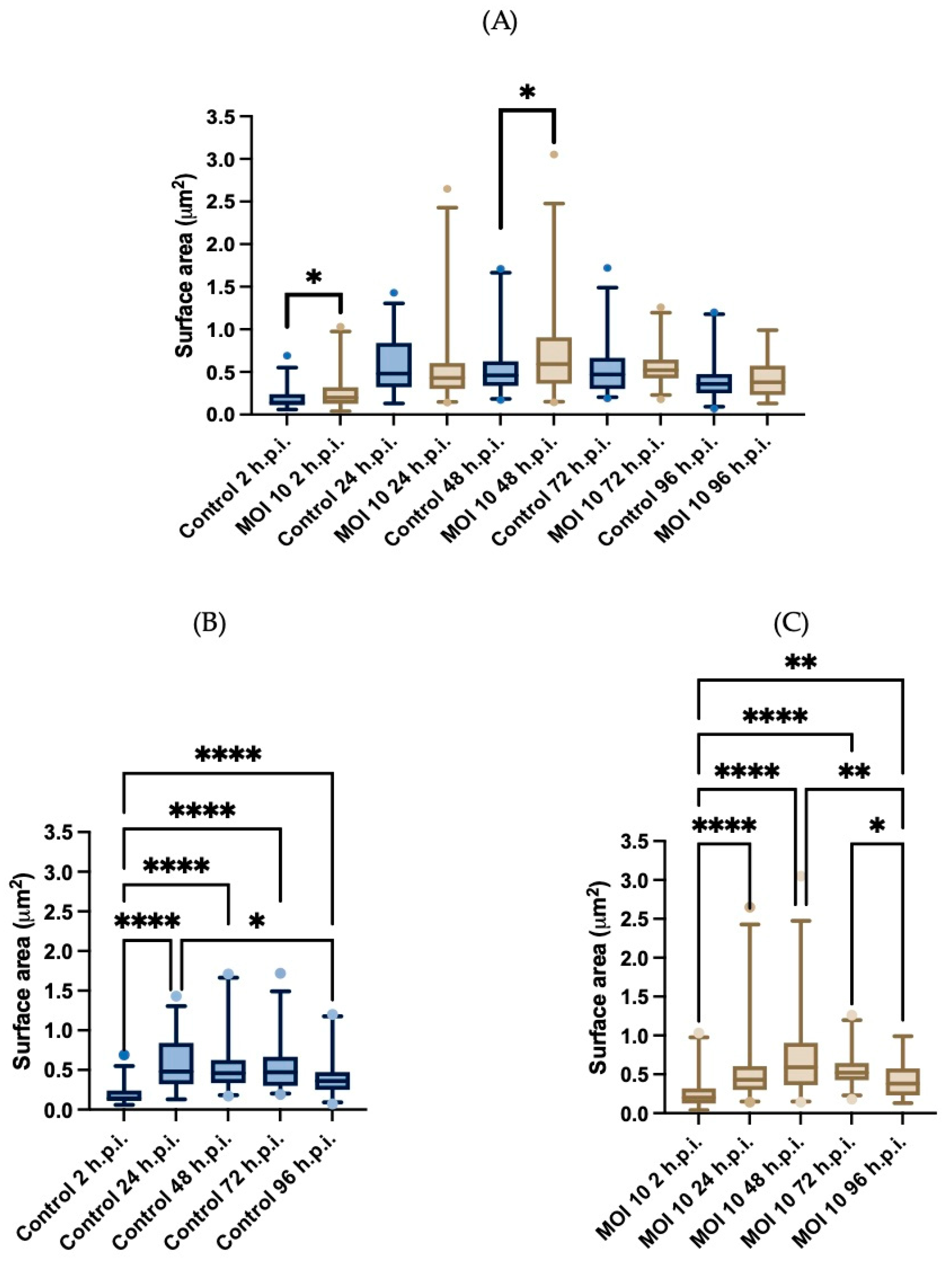
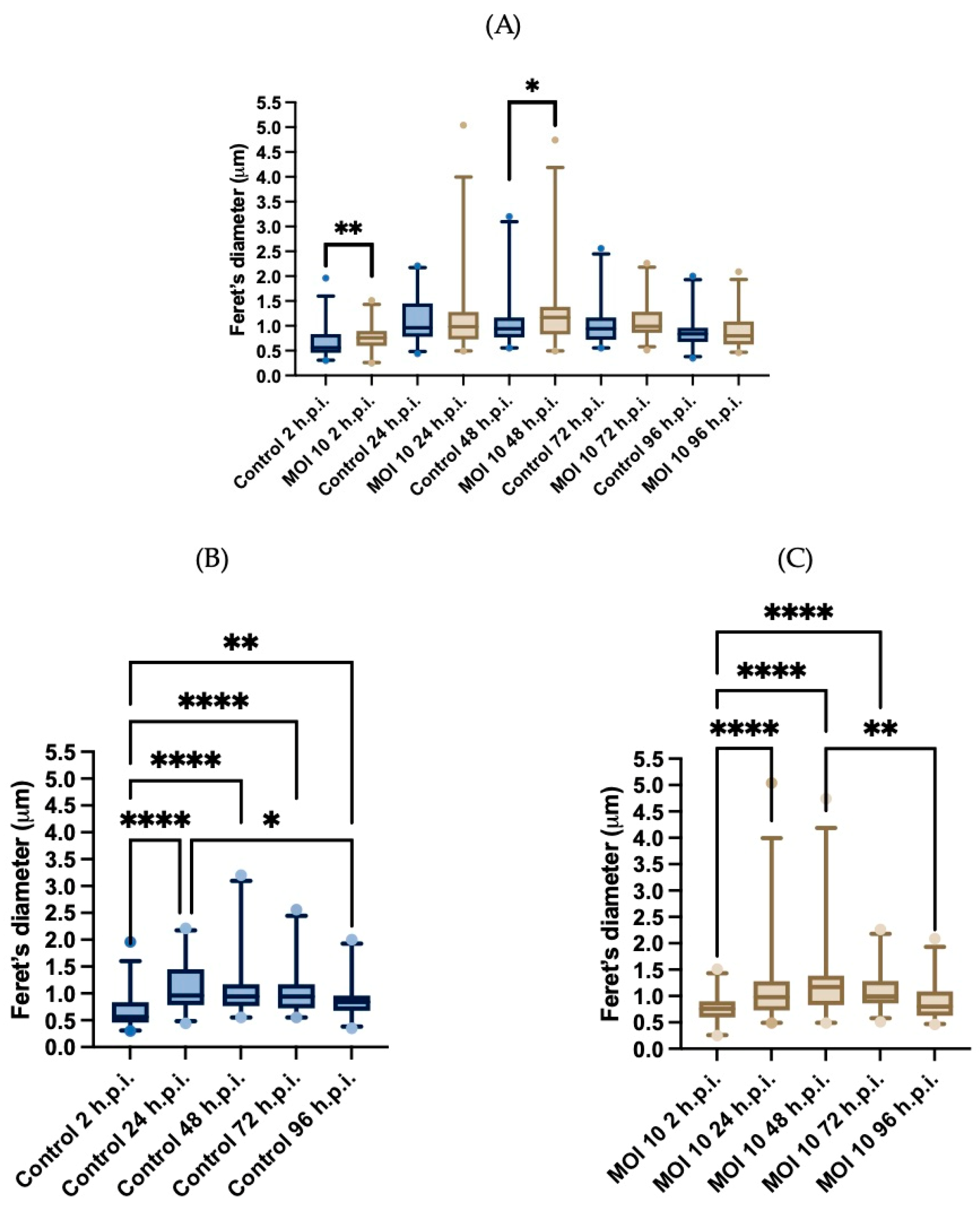
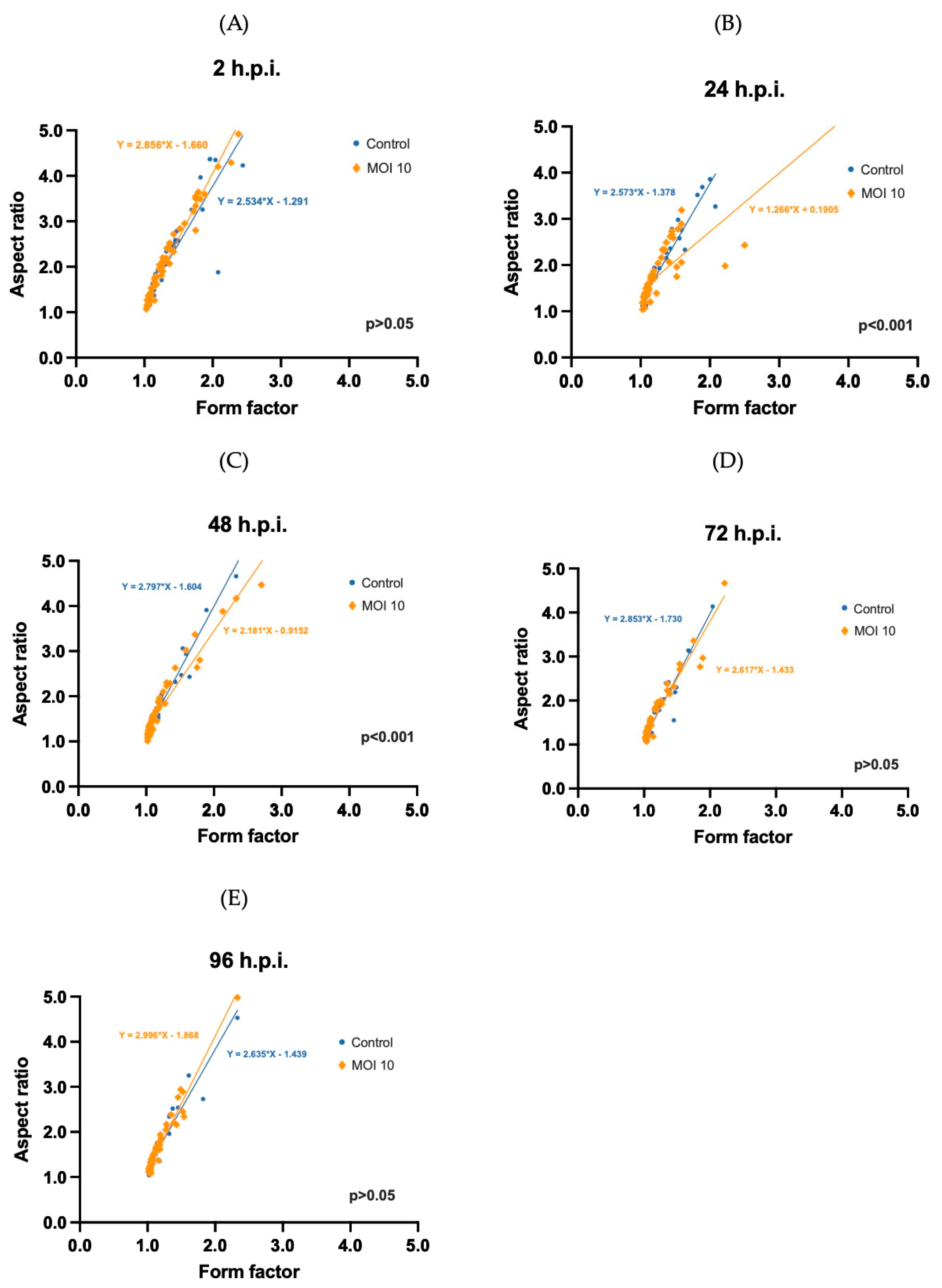
3.3. Morphometric Analysis of RSV ON1-Infected Mitochondria in the HEp-2 Cells Line
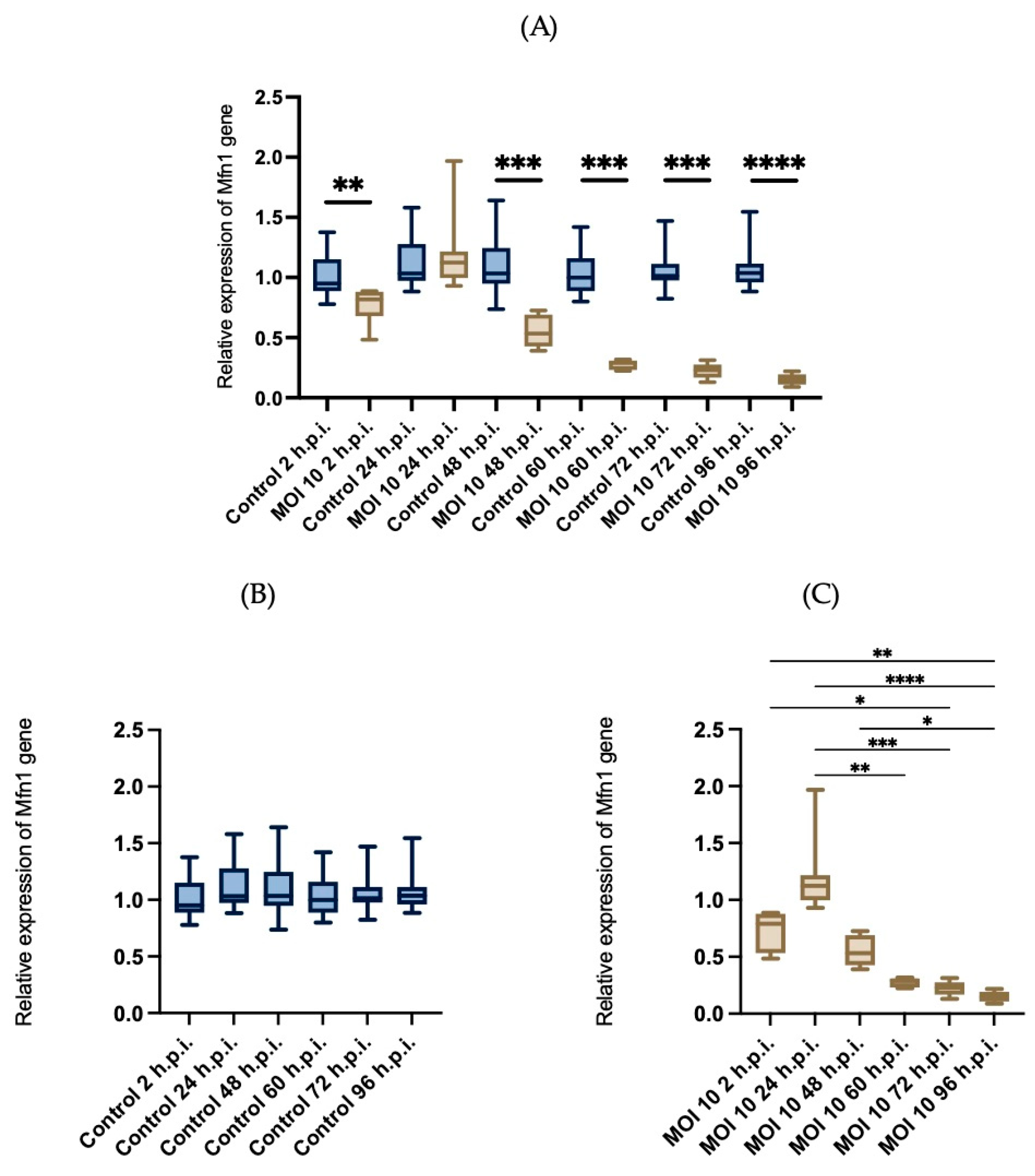
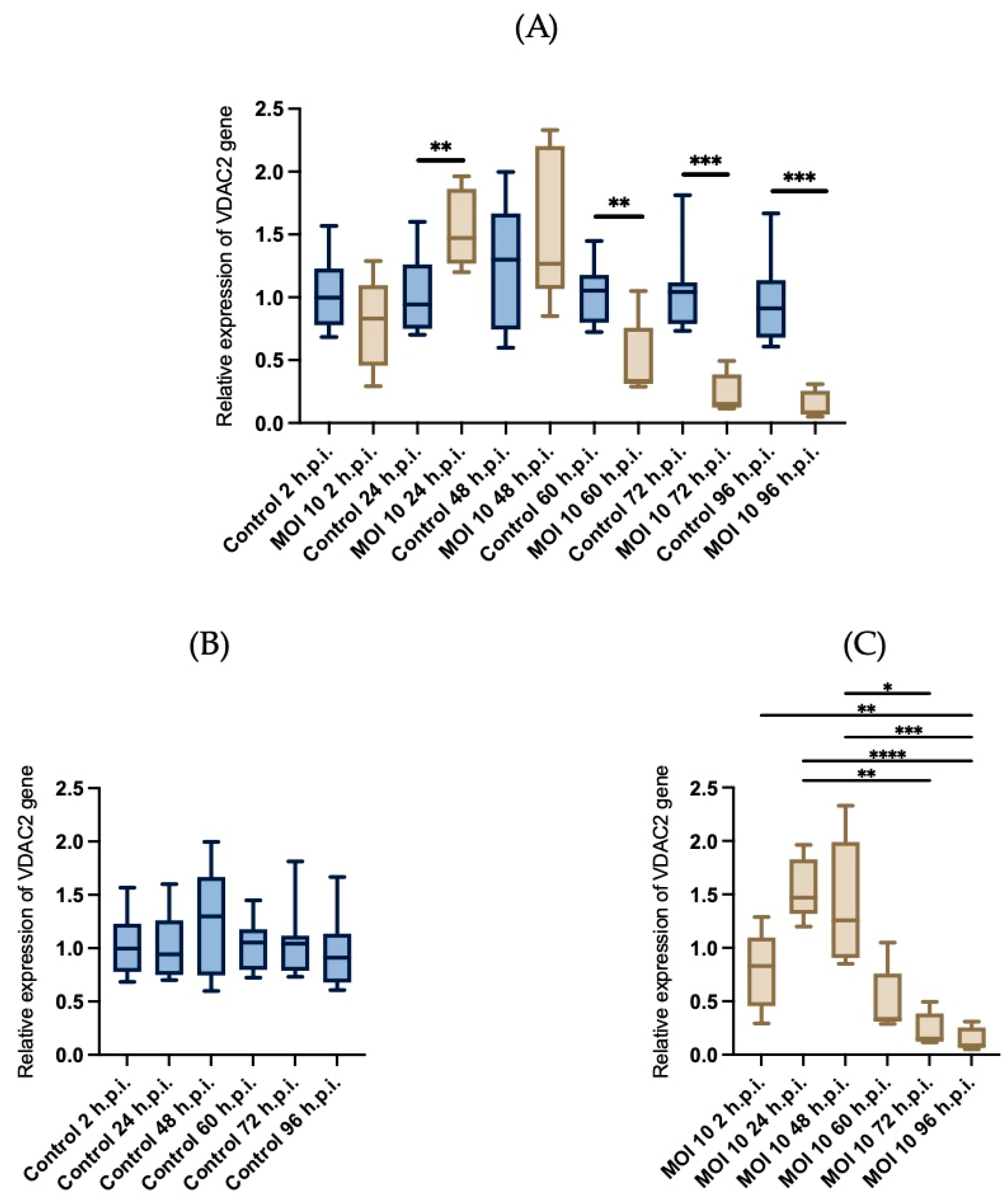
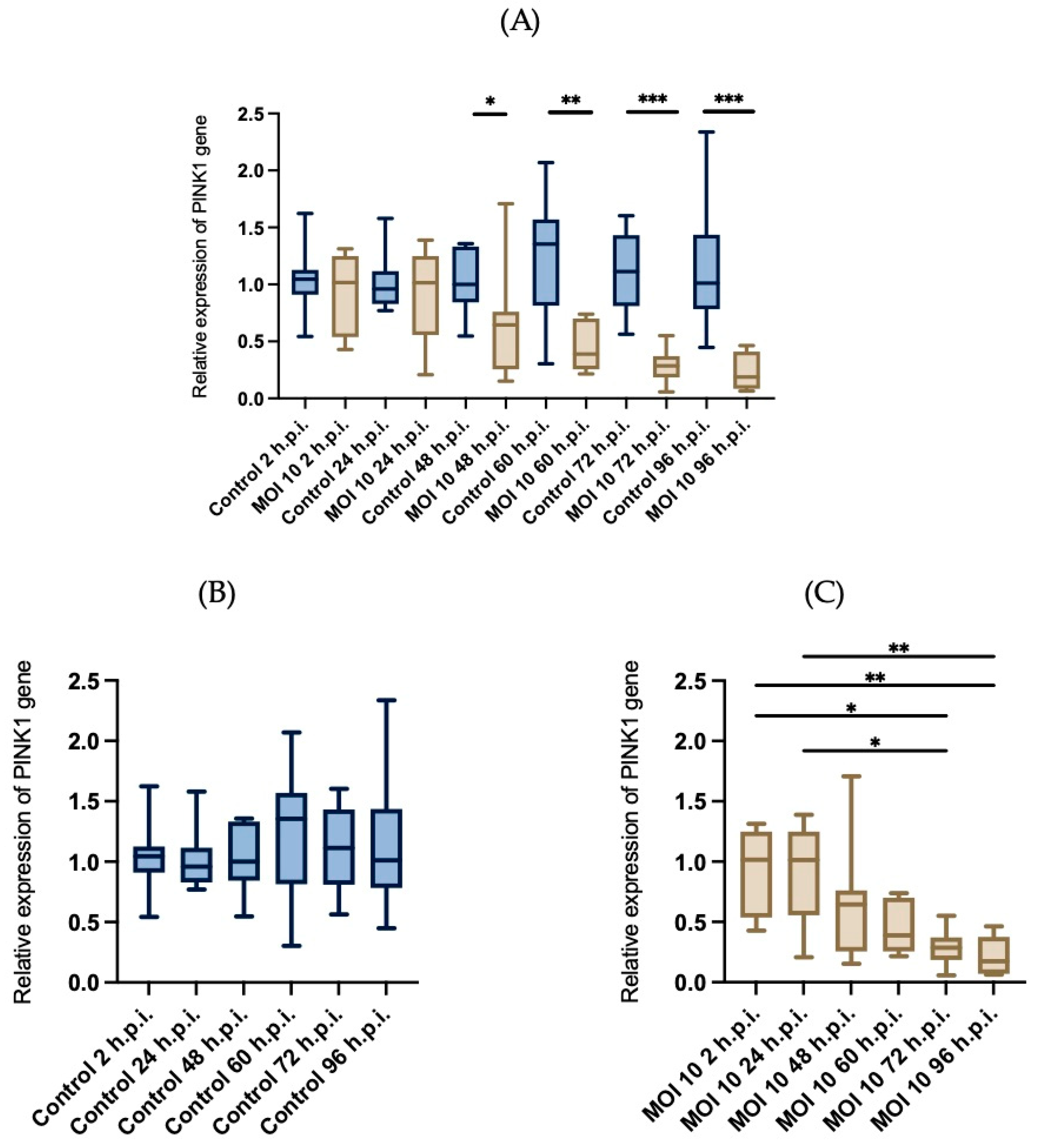
3.4. Effects of RSV ON1 Infection on HEp-2 Cell Line Mitochondrial Function Genes Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.R.; Spriggs, M.K.; Olmsted, R.A.; Collins, P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 5625–5629. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- García, O.; Martín, M.; Dopazo, J.; Arbiza, J.; Frabasile, S.; Russi, J.; Hortal, M.; Perez-Breña, P.; Martínez, I.; García-Barreno, B. Evolutionary pattern of human respiratory syncytial virus (subgroup A): Cocirculating lineages and correlation of genetic and antigenic changes in the G glycoprotein. J. Virol. 1994, 68, 5448–5459. [Google Scholar] [CrossRef] [Green Version]
- Trento, A.; Galiano, M.; Videla, C.; Carballal, G.; García-Barreno, B.; Melero, J.A.; Palomo, C. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J. Gen. Virol. 2003, 84, 3115–3120. [Google Scholar] [CrossRef]
- Muñoz-Escalante, J.C.; Comas-García, A.; Bernal-Silva, S.; Noyola, D.E. Respiratory syncytial virus B sequence analysis reveals a novel early genotype. Sci. Rep. 2021, 11, 3452. [Google Scholar] [CrossRef]
- Eshaghi, A.; Duvvuri, V.R.; Lai, R.; Nadarajah, J.T.; Li, A.; Patel, S.N.; Low, D.E.; Gubbay, J.B. Genetic Variability of Human Respiratory Syncytial Virus A Strains Circulating in Ontario: A Novel Genotype with a 72 Nucleotide G Gene Duplication. PLoS ONE 2012, 7, e32807. [Google Scholar] [CrossRef] [Green Version]
- Comas-García, A.; Noyola, D.E.; Cadena-Mota, S.; Rico-Hernández, M.; Bernal-Silva, S. Respiratory Syncytial Virus-A ON1 Genotype Emergence in Central Mexico in 2009 and Evidence of Multiple Duplication Events. J. Infect. Dis. 2018, 217, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Cantú-Flores, K.; Rivera-Alfaro, G.; Muñoz-Escalante, J.C.; Noyola, D.E. Global distribution of respiratory syncytial virus A and B infections: A systematic review. Pathog. Glob. Health 2022, 116, 398–409. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- de Armas-Rillo, L.; Valera, M.-S.; Marrero-Hernández, S.; Valenzuela-Fernández, A. Membrane dynamics associated with viral infection. Rev. Med. Virol. 2016, 26, 146–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowton, V.M.; Fearns, R. Evidence that the respiratory syncytial virus polymerase is recruited to nucleotides 1 to 11 at the 3′ end of the nucleocapsid and can scan to access internal signals. J. Virol. 2005, 79, 11311–11322. [Google Scholar] [CrossRef] [Green Version]
- Ghildyal, R.; Li, D.; Peroulis, I.; Shields, B.; Bardin, P.G.; Teng, M.N.; Collins, P.L.; Meanger, J.; Mills, J. Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J. Gen. Virol. 2005, 86 Pt 7, 1879–1884. [Google Scholar] [CrossRef]
- Ghildyal, R.; Mills, J.; Murray, M.; Vardaxis, N.; Meanger, J. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J. Gen. Virol. 2022, 83 Pt 4, 753–757. [Google Scholar] [CrossRef]
- Reimers, K.; Buchholz, K.; Werchau, H. Respiratory syncytial virus M2-1 protein induces the activation of nuclear factor kappa B. Virology 2005, 331, 260–268. [Google Scholar] [CrossRef]
- Li, D.; Jans, D.A.; Bardin, P.G.; Meanger, J.; Mills, J.; Ghildyal, R. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J. Virol. 2008, 82, 8863–8870. [Google Scholar] [CrossRef] [Green Version]
- Li, H.M.; Ghildyal, R.; Hu, M.; Tran, K.C.; Starrs, L.M.; Mills, J.; Teng, M.N.; Jans, D.A. Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells. Cells 2021, 10, 2786. [Google Scholar] [CrossRef]
- Bossert, B.; Marozin, S.; Conzelmann, K.K. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J. Virol. 2003, 77, 8661–8668. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.; Lynch, O.T.; Suessmuth, Y.; Qian, P.; Boyd, C.R.; Burrows, J.F.; Buick, R.; Stevenson, N.J.; Touzelet, O.; Gadina, M.; et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J. Virol. 2007, 81, 3428–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E., Jr.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, H.M.; Bogoyevitch, M.A.; Jans, D.A. Mitochondrial protein p32/HAPB1/gC1qR/C1qbp is required for efficient respiratory syncytial virus production. Biochem. Biophys. Res. Commun. 2017, 489, 460–465. [Google Scholar] [CrossRef]
- Kim, S.-J.; Ahn, D.-G.; Syed, G.H.; Siddiqui, A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 2017, 41, 21–27. [Google Scholar] [CrossRef]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Kim, S.-J.; Syed, G.H.; Siddiqui, A. Hepatitis C Virus Induces the Mitochondrial Translocation of Parkin and Subsequent Mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.-M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [Green Version]
- Kipper, S.; Hamad, S.; Caly, L.; Avrahami, D.; Bacharach, E.; Jans, D.A.; Gerber, D.; Bajorek, M. New host factors important for respiratory syncytial virus (RSV) replication revealed by a novel microfluidics screen for interactors of matrix (M) protein. Mol. Cell. Proteom. 2015, 14, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Munday, D.C.; Howell, G.; Barr, J.N.; Hiscox, J.A. Proteomic analysis of mitochondria in respiratory epithelial cells infected with human respiratory syncytial virus and functional implications for virus and cell biology. J. Pharm. Pharmacol. 2015, 67, 300–318. [Google Scholar] [CrossRef]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. eLife 2019, 8, e42448. [Google Scholar] [CrossRef] [PubMed]
- Comas-García, A.; López-Pacheco, C.P.; García-Zepeda, E.A.; Soldevila, G.; Ramos-Martínez, P.; Ramos-Castañeda, J. Neonatal respiratory syncytial virus infection has an effect on lung inflammation and the CD4+ CD25+ T cell subpopulation during ovalbumin sensitization in adult mice. Clin. Exp. Immunol. 2016, 185, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Lombardía, L.; Herranz, C.; García-Barreno, B.; Domínguez, O.; Melero, J.A. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009, 388, 31–41. [Google Scholar] [CrossRef] [Green Version]
- McKimm-Breschkin, J.L. A simplified plaque assay for respiratory syncytial virus—Direct visualization of plaques without immunostaining. J. Virol. Methods 2004, 120, 113–117. [Google Scholar] [CrossRef]
- King, T.; Mejias, A.; Ramilo, O.; Peeples, M.E. The larger attachment glycoprotein of respiratory syncytial virus produced in primary human bronchial epithelial cultures reduces infectivity for cell lines. PLoS Pathog. 2021, 17, e1009469. [Google Scholar] [CrossRef] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef] [PubMed]
- Bossert, B.; Conzelmann, K.K. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: A chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J. Virol. 2002, 76, 4287–4293. [Google Scholar] [CrossRef] [Green Version]
- Rayaprolu, V.; Ramsey, J.; Wang, J.C.; Mukhopadhyay, S. Alphavirus Purification Using Low-speed Spin Centrifugation. Bio-Protocol 2018, 8, e2772. [Google Scholar] [CrossRef]
- Tobin, G.J.; Nagashima, K.; Gonda, M.A. Immunologic and Ultrastructural Characterization of HIV Pseudovirions Containing Gag and Env Precursor Proteins Engineered in Insect Cells. Methods 1996, 10, 208–218. [Google Scholar] [CrossRef]
- Crouser, E.D.; Julian, M.W.; Dorinsky, P.M. Ileal VO(2)-O(2) alterations induced by endotoxin correlate with severity of mitochondrial injury. Am. J. Respir. Crit. Care Med. 1999, 160, 1347–1353. [Google Scholar] [CrossRef]
- Joshi, M.S.; Crouser, E.D.; Julian, M.W.; Schanbacher, B.L.; Bauer, J.A. Digital imaging analysis for the study of endotoxin-induced mitochondrial ultrastructure injury. Anal. Cell. Pathol. 2000, 21, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chiok, K.; Pokharel, S.M.; Mohanty, I.; Miller, L.G.; Gao, S.J.; Haas, A.L.; Tran, K.C.; Teng, M.N.; Bose, S. Human Respiratory Syncytial Virus NS2 Protein Induces Autophagy by Modulating Beclin1 Protein Stabilization and ISGylation. mBio 2022, 13, e0352821. [Google Scholar] [CrossRef] [PubMed]
- Raiden, S.; Sananez, I.; Remes-Lenicov, F.; Pandolfi, J.; Romero, C.; De Lillo, L.; Ceballos, A.; Geffner, J.; Arruvito, L. Respiratory Syncytial Virus (RSV) Infects CD4+ T Cells: Frequency of Circulating CD4+ RSV+ T Cells as a Marker of Disease Severity in Young Children. J. Infect. Dis. 2017, 215, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of Host Cell Mitochondria by RSV to Favor Virus Production depends on the Inhibition of Mitochondrial Complex I and ROS Generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Kulkarni, H.; Radhakrishnan, P.; Rutman, A.; Bankart, M.J.; Williams, G.; Hirst, R.A.; Easton, A.J.; Andrew, P.W.; O’Callaghan, C. Ciliary dyskinesia is an early feature of respiratory syncytial virus infection. Eur. Respir. J. 2014, 43, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Hernández, E.I.; Comas-García, M.; Coronado-Ipiña, M.A.; Colunga-Saucedo, M.; González Sánchez, H.M.; Castillo, C.G. Astrocytes derived from neural progenitor cells are susceptible to Zika virus infection. PLoS ONE 2023, 18, e0283429. [Google Scholar] [CrossRef] [PubMed]
- Colunga-Saucedo, M.; Rubio-Hernandez, E.I.; Coronado-Ipiña, M.A.; Rosales-Mendoza, S.; Castillo, C.G.; Comas-Garcia, M. Construction of a Chikungunya Virus, Replicon, and Helper Plasmids for Transfection of Mammalian Cells. Viruses 2022, 15, 132. [Google Scholar] [CrossRef]
- Zhong, G.Y. Ultrastructural changes in middle ear and cochlea caused by respiratory syncytial virus in guinea pigs. Zhonghua Er Bi Yan Hou Ke Za Zhi 1990, 25, 213–215, 255. [Google Scholar]
- Cortese, M.; Lee, J.-Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e5. [Google Scholar] [CrossRef]
- Sato, H.; Hoshi, M.; Ikeda, F.; Fujiyuki, T.; Yoneda, M.; Kai, C. Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLoS Pathog. 2021, 17, e1009841. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pila-Castellanos, I.; Molino, D.; McKellar, J.; Lines, L.; Da Graca, J.; Tauziet, M.; Chanteloup, L.; Mikaelian, I.; Meyniel-Schicklin, L.; Codogno, P.; et al. Mitochondrial morphodynamic alteration induced by influenza virus infection as a new antiviral strategy. PLoS Pathog. 2021, 17, e1009340. [Google Scholar] [CrossRef]
- Brandt, T.; Cavellini, L.; Kühlbrandt, W.; Cohen, M.M. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. eLife 2016, 5, e14618. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Liou, Y.C. Functions of outer mitochondrial membrane proteins: Mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2020, 28, 827–842. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptor and RIG-1-like Receptor Signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2009, 11, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Onoguchi, K.; Onomoto, K.; Takamatsu, S.; Jogi, M.; Takemura, A.; Morimoto, S.; Julkunen, I.; Namiki, H.; Yoneyama, M.; Fujita, T. Virus-Infection or 5′ppp-RNA Activates Antiviral Signal through Redistribution of IPS-1 Mediated by MFN1. PLoS Pathog. 2010, 6, e1001012. [Google Scholar] [CrossRef] [Green Version]
- Elesela, S.; Morris, S.B.; Narayanan, S.; Kumar, S.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 regulates mitochondrial function and immune homeostasis in respiratory syncytial virus infected dendritic cells. PLoS Pathog. 2020, 16, e1008319. [Google Scholar] [CrossRef] [Green Version]
- Oanh, N.T.K.; Park, Y.-Y.; Cho, H. Mitochondria elongation is mediated through SIRT1-mediated MFN1 stabilization. Cell. Signal. 2017, 38, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Liang, J.-J.; Li, J.-K.; Lee, Y.-L.; Chang, B.-L.; Su, C.-I.; Huang, W.-J.; Lai, M.M.C.; Lin, Y.-L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, R.P.; Buzhynskyy, N.; Prima, V.; Sturgis, J.N.; Scheuring, S. Supramolecular Assembly of VDAC in Native Mitochondrial Outer Membranes. J. Mol. Biol. 2007, 369, 413–418. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Keinan, N.; Zaid, H. Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 2008, 40, 183–191. [Google Scholar] [CrossRef]
- Zamarin, D.; García-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza Virus PB1-F2 Protein Induces Cell Death through Mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, K.; Zeng, S.; Zou, L.; Li, X.; Xu, C.; Li, B.; Liu, X.; Li, Z.; Zhu, W.; et al. The Role of Mitophagy in Viral Infection. Cells 2022, 11, 711. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, R.; Zhang, Z.; Liu, Q.; Zhang, Y.; Wang, Q.; Yuan, H. Mitochondrial Protein PINK1 Positively Regulates RLR Signaling. Front. Immunol. 2019, 10, 1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhvalova, M.S.; Prince, G.A.; Blanco, J.C.G. Respiratory Syncytial Virus Infects and Abortively Replicates in the Lungs in Spite of Preexisting Immunity. J. Virol. 2007, 81, 9443–9450. [Google Scholar] [CrossRef] [Green Version]
- Piedra, F.-A.; Qiu, X.; Teng, M.N.; Avadhanula, V.; Machado, A.A.; Kim, D.-K.; Hixson, J.; Bahl, J.; Piedra, P.A. Non-gradient and genotype-dependent patterns of RSV gene expression. PLoS ONE 2020, 15, e0227558. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer’s Sequence (5′-3′) |
|---|---|
| PINK1 forward | GAGTATGGAGCAGTCACTTACAG |
| PINK1 reverse | CAGCACATCAGGGTAGTCG |
| Mfn1 forward | ACCTGTTTCTCCACTGAAGC |
| Mfn1 reverse | TGGCTATTCGATCAAGTTCCG |
| VDAC2 forward | TGTCTTTGGTTATGAGGGCTG |
| VDAC2 reverse | CCTCCAAATTCTGTCCCATCG |
| GAPDH forward | CAAGGCTGAGAACGGGAAGC |
| GAPDH reverse | AGGGGGCAGAGATGATGACC |
| MCVF | GGCAAATATGGAAACATACGTGAA |
| MCVR | TCTTTTTCTAGGACATTGTAYTGAACA |
| Injury Stage | Characteristics of Mitochondria |
|---|---|
| Class I | Mitochondria are considered normal. The cristae are densely packed and longitudinal, and the matrix is electron dense. |
| Class II | Mitochondria in this class have rounded or asymmetrical cristae. The cristae’s direction, narrowness, or regular distribution have been lost. |
| Class III | Mitochondria vary in size and form, with discontinuous membranes, fragmented cristae, and swollen and electro lucent matrix. |
| Class IV | In this class, the mitochondrial double membrane is no longer complete, and the cristae are disrupted and dispersed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara-Hernandez, I.; Muñoz-Escalante, J.C.; Bernal-Silva, S.; Noyola, D.E.; Wong-Chew, R.M.; Comas-García, A.; Comas-Garcia, M. Ultrastructural and Functional Characterization of Mitochondrial Dynamics Induced by Human Respiratory Syncytial Virus Infection in HEp-2 Cells. Viruses 2023, 15, 1518. https://doi.org/10.3390/v15071518
Lara-Hernandez I, Muñoz-Escalante JC, Bernal-Silva S, Noyola DE, Wong-Chew RM, Comas-García A, Comas-Garcia M. Ultrastructural and Functional Characterization of Mitochondrial Dynamics Induced by Human Respiratory Syncytial Virus Infection in HEp-2 Cells. Viruses. 2023; 15(7):1518. https://doi.org/10.3390/v15071518
Chicago/Turabian StyleLara-Hernandez, Ignacio, Juan Carlos Muñoz-Escalante, Sofía Bernal-Silva, Daniel E. Noyola, Rosa María Wong-Chew, Andreu Comas-García, and Mauricio Comas-Garcia. 2023. "Ultrastructural and Functional Characterization of Mitochondrial Dynamics Induced by Human Respiratory Syncytial Virus Infection in HEp-2 Cells" Viruses 15, no. 7: 1518. https://doi.org/10.3390/v15071518
APA StyleLara-Hernandez, I., Muñoz-Escalante, J. C., Bernal-Silva, S., Noyola, D. E., Wong-Chew, R. M., Comas-García, A., & Comas-Garcia, M. (2023). Ultrastructural and Functional Characterization of Mitochondrial Dynamics Induced by Human Respiratory Syncytial Virus Infection in HEp-2 Cells. Viruses, 15(7), 1518. https://doi.org/10.3390/v15071518