
BMS-265246, a Cyclin-Dependent Kinase Inhibitor, Inhibits the Infection of Herpes Simplex Virus Type 1


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Infection
2.2. Compounds
2.3. Antibodies
2.4. Cell Viability Assay
2.5. Plaque Assay
2.6. Antiviral Activity Assays
2.7. Time-of-Addition Assay
2.8. Indirect Immunofluorescence Assay
2.9. Virus Attachment Assay
2.10. Virus Entry Assay
2.11. Western Blot
2.12. Quantitative Real-Time PCR of Viral mRNA and DNA
2.13. Statistical Analyses
3. Results
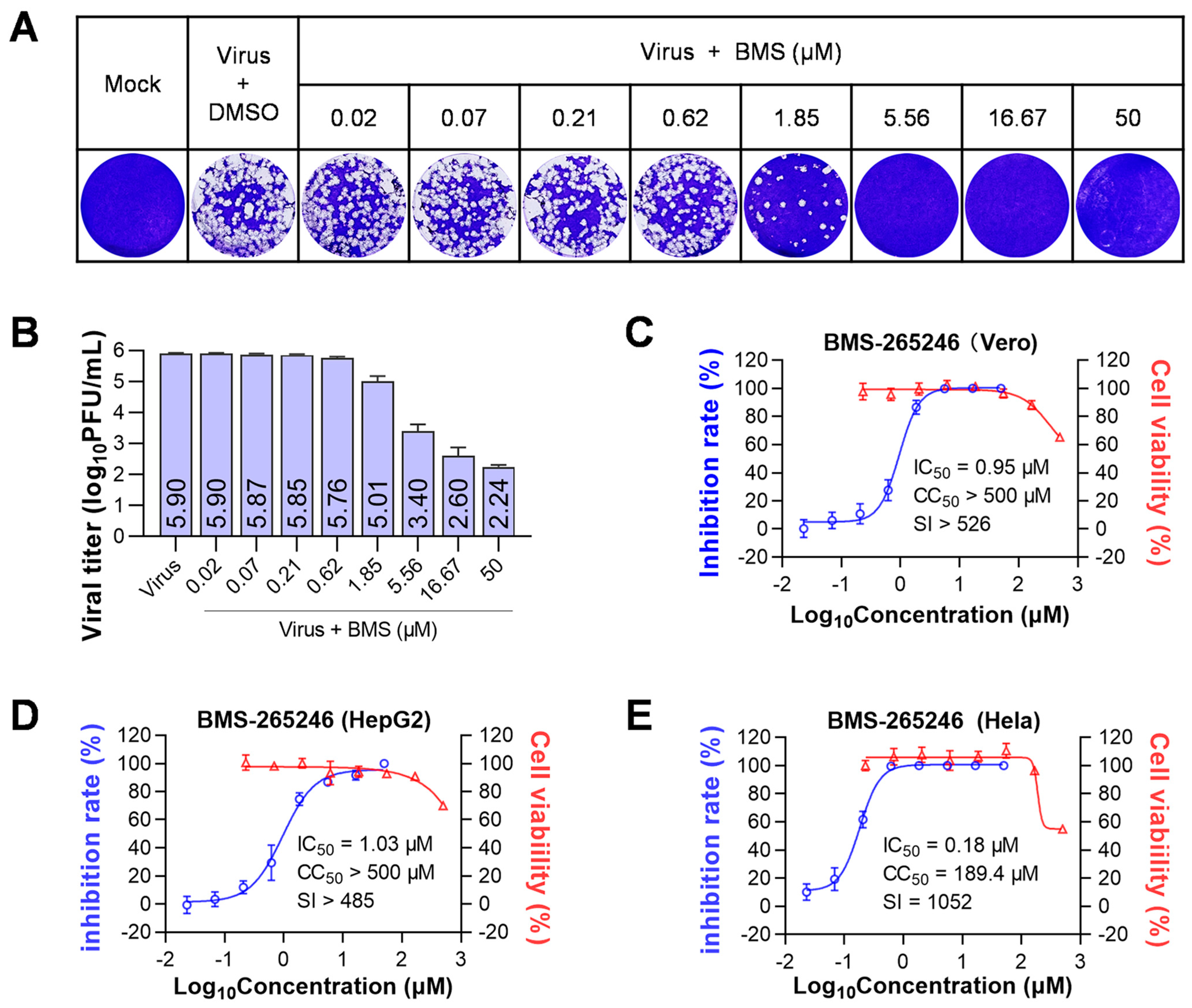
3.1. BMS Was Identified to Inhibit the Replication of HSV-1 and HSV-2 In Vitro
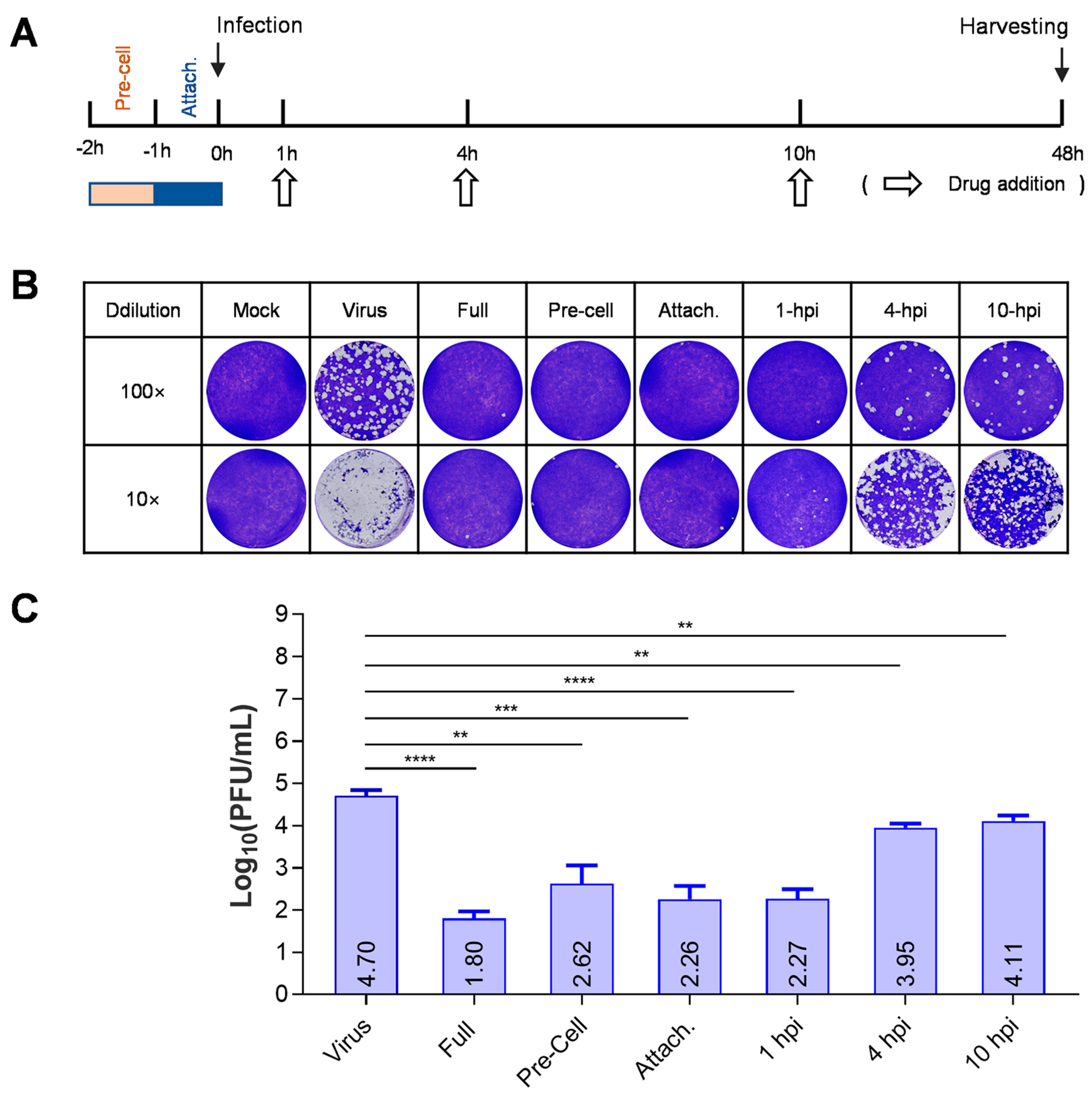
3.2. BMS Inhibits the Early Stage of HSV-1 Replication
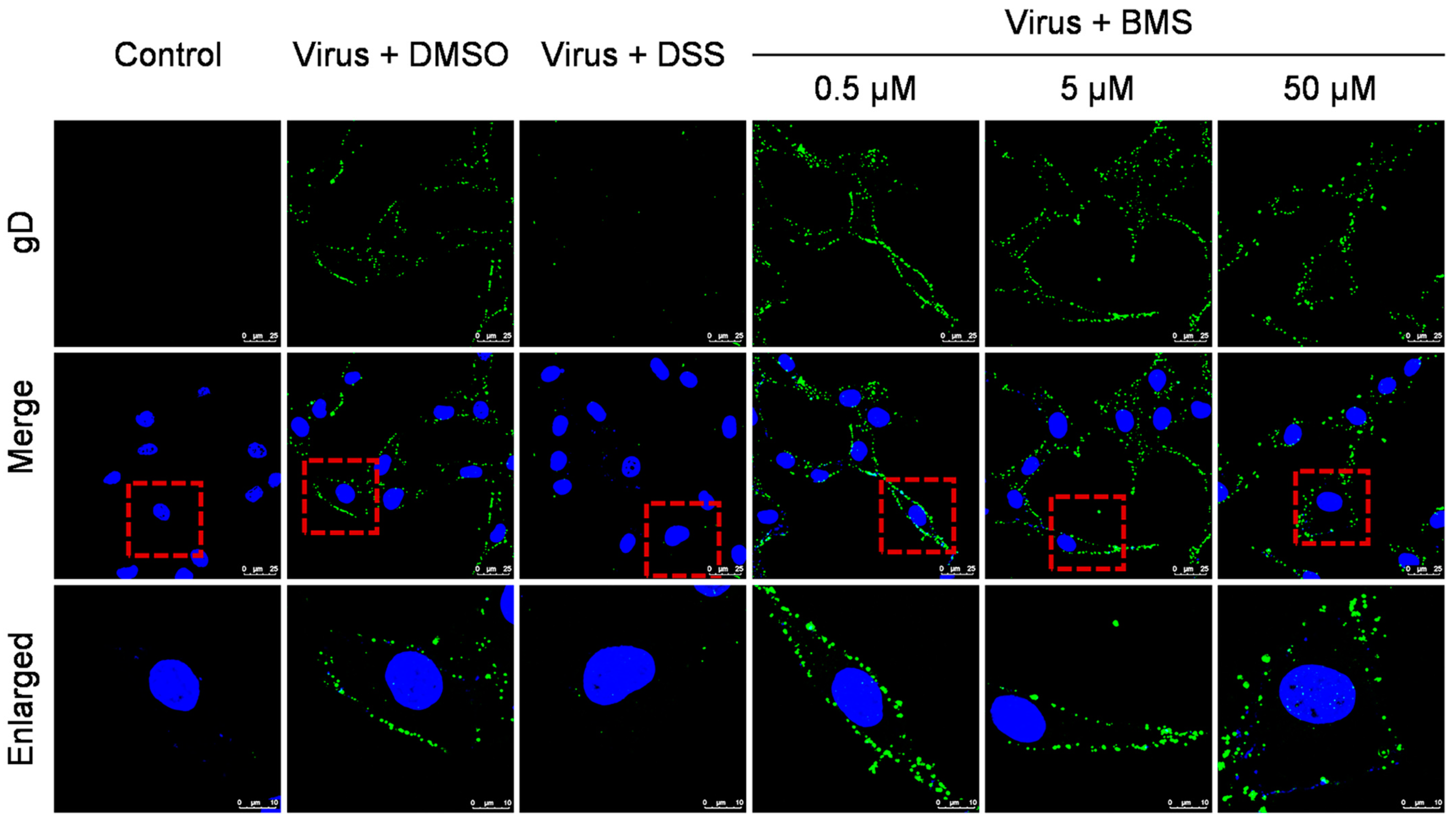
3.3. BMS Does Not Inhibit HSV-1 Attachment to Host Cells
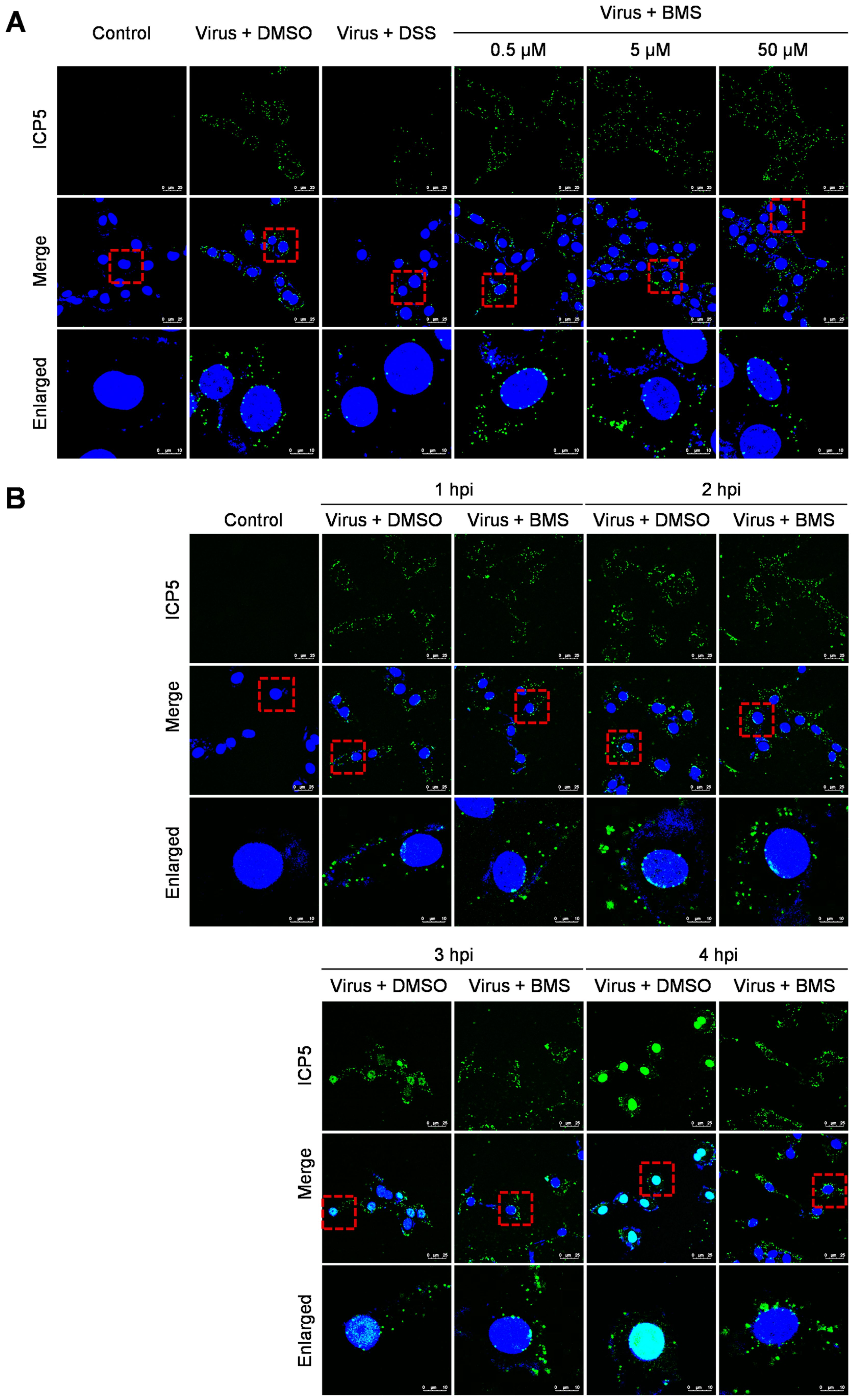
3.4. BMS Does Not Inhibit the Entry of HSV-1 into Host Cells
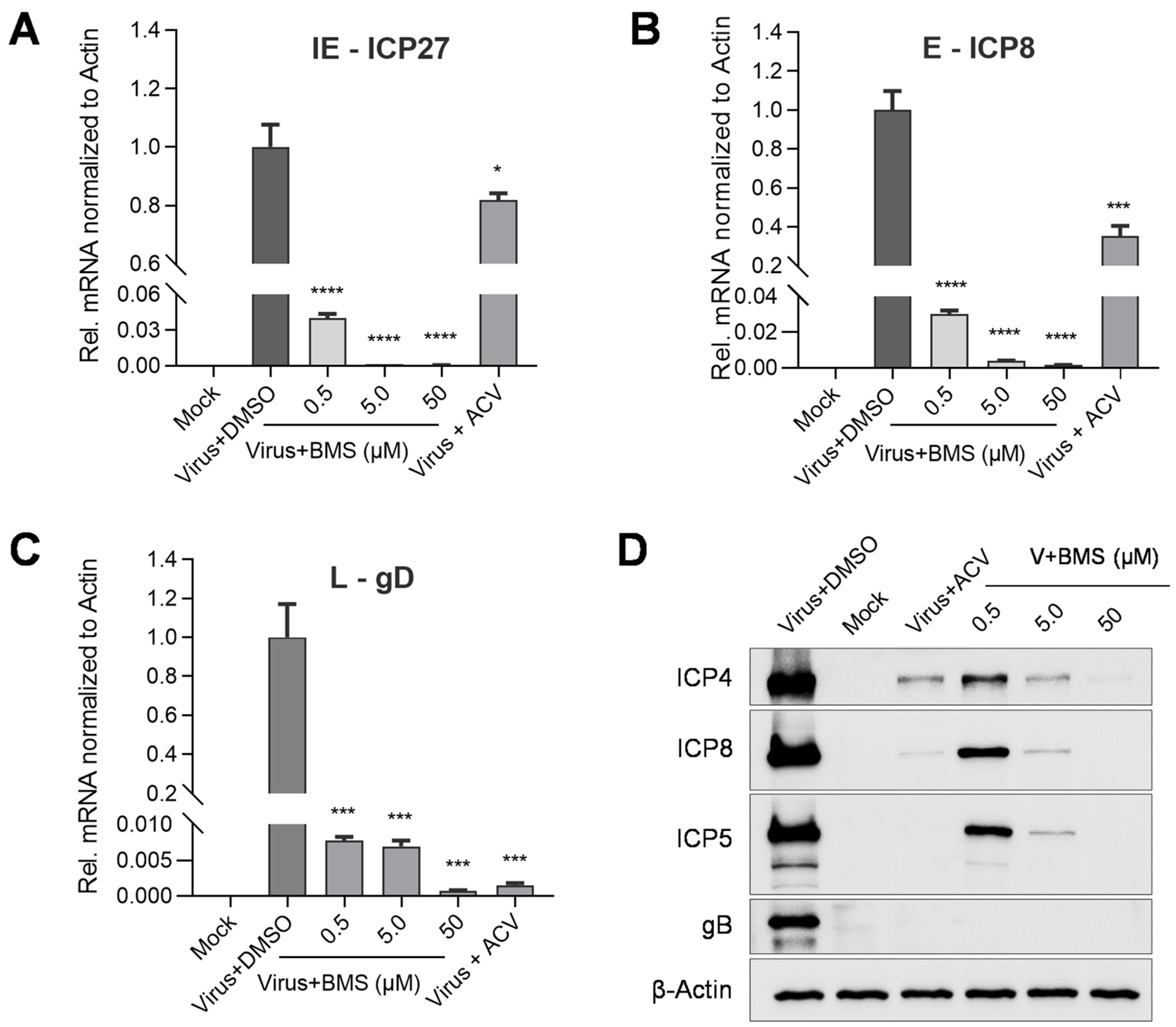
3.5. The Expression of HSV-1 IE, E, and L Genes Are Affected by BMS Treatment
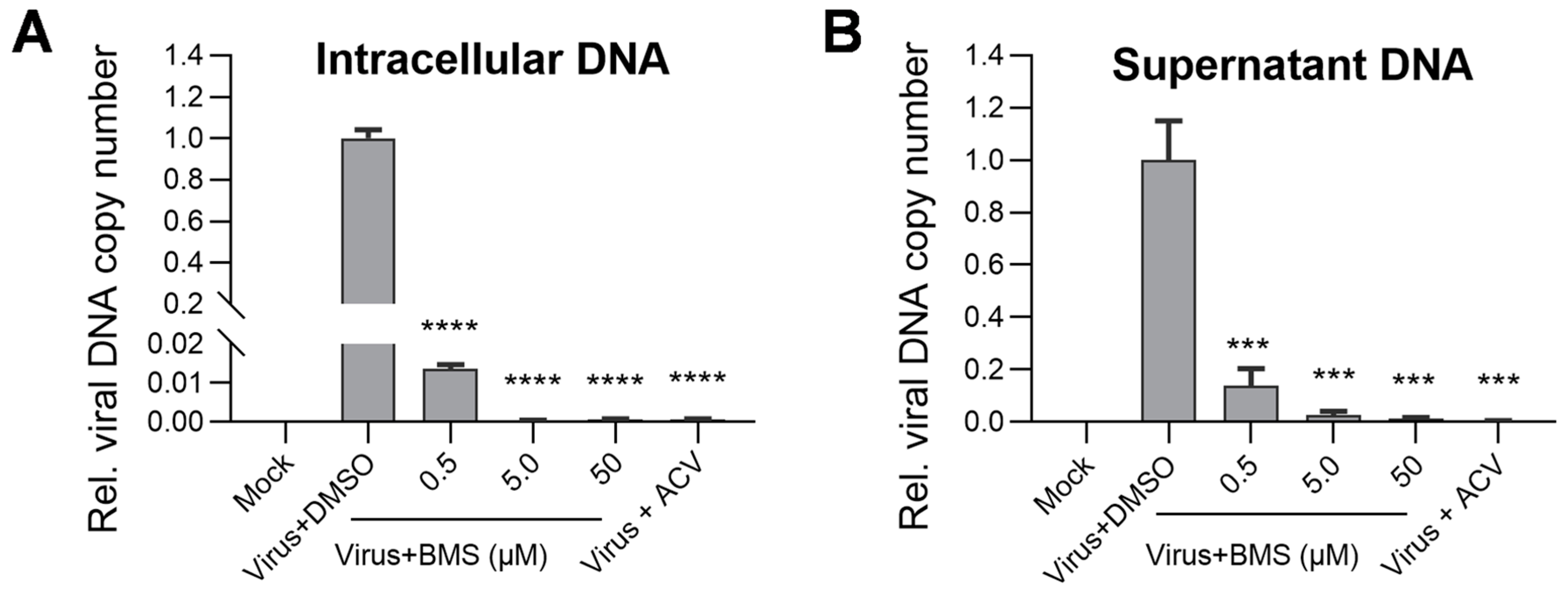
3.6. The Intracellular and Virion-Associated HSV-1 Viral DNA Levels Are Decreased upon BMS Treatment
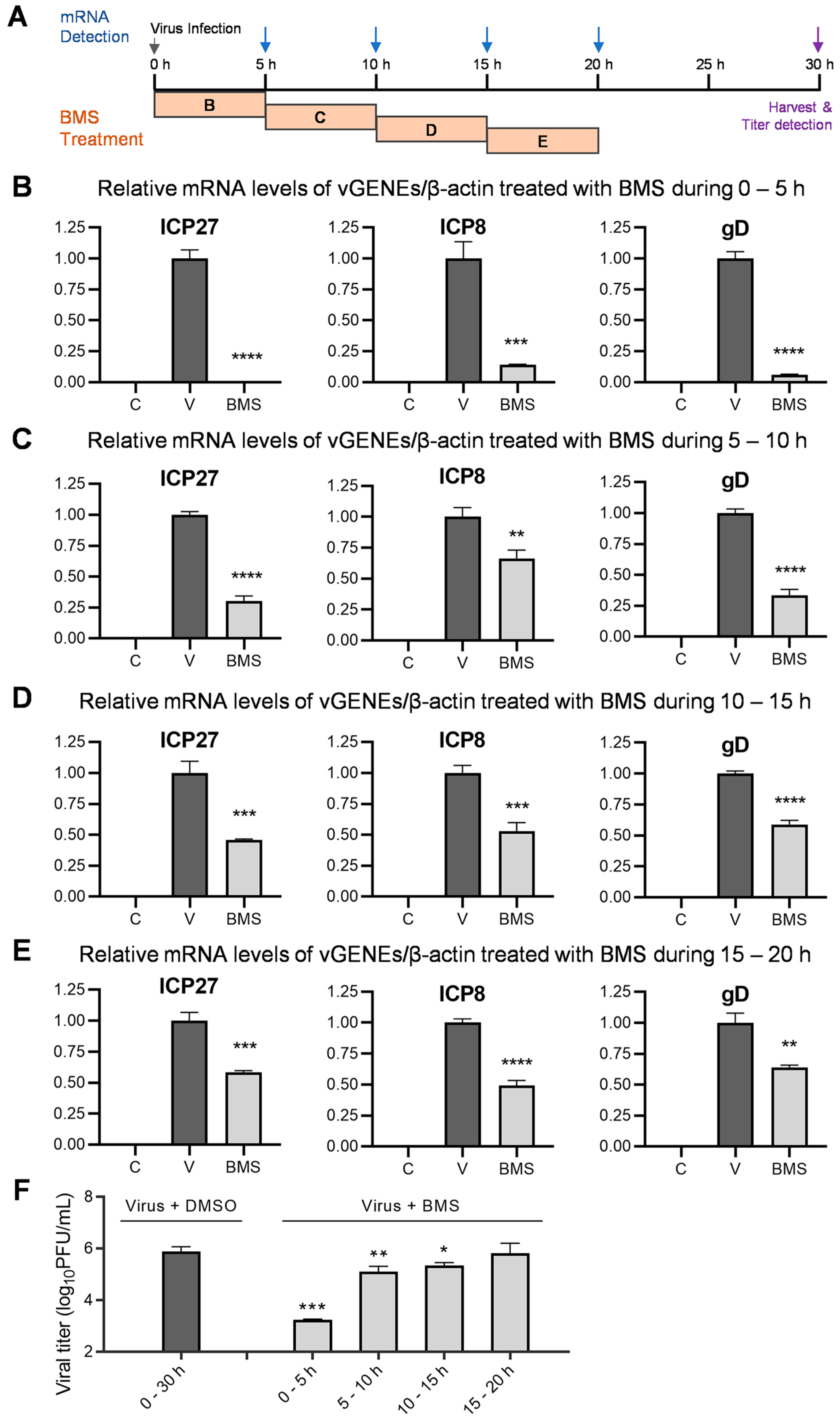
3.7. BMS Suppresses the Transcription of IE, E, and L Genes of HSV-1 during Multiple Stages of Virus Replication
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Herpes Simplex Virus. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 5 April 2023).
- Oakley, C.; Epstein, J.; Sherlock, C. Reactivation of oral herpes simplex virus: Implications for clinical management of herpes simplex virus recurrence during radiotherapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 1997, 84, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Greenberg, M.S. Chronic oral herpes simplex virus infection in immunocompromised patients. Oral Surg. Oral Med. Oral Pathol. 1985, 59, 465–471. [Google Scholar] [CrossRef]
- Prétet, J.-L.; Pelletier, L.; Bernard, B.; Coumes-Marquet, S.; Kantelip, B.; Mougin, C. Apoptosis participates to liver damage in HSV-induced fulminant hepatitis. Apoptosis 2003, 8, 655–663. [Google Scholar] [CrossRef]
- Price, N.B.; Wood, K.E. Distinguishing Features Common to Dual Fatal Herpes Simplex Virus Infections That Occur in Both a Pregnant Woman and Her Newborn Infant. Viruses 2021, 13, 2542. [Google Scholar] [CrossRef] [PubMed]
- Samies, N.L.; James, S.H.; Kimberlin, D.W. Neonatal Herpes Simplex Virus Disease: Updates and Continued Challenges. Clin. Perinatol. 2021, 48, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Field, H.J.; Hodge, R.A.V. Recent developments in anti-herpesvirus drugs. Br. Med. Bull. 2013, 106, 213–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, R.A.V.; Field, H.J. Antiviral Agents for Herpes Simplex Virus. Adv. Pharmacol. 2013, 67, 1–38. [Google Scholar] [CrossRef]
- Shiraki, K.; Yasumoto, S.; Toyama, N.; Fukuda, H. Amenamevir, a Helicase-Primase Inhibitor, for the Optimal Treatment of Herpes Zoster. Viruses 2021, 13, 1547. [Google Scholar] [CrossRef]
- Morfin, F.; Thouvenot, D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 2003, 26, 29–37. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Zhu, Q.; Zhou, R.; Liu, J.; Peng, T. Identification and characterization of acyclovir-resistant clinical HSV-1 isolates from children. J. Clin. Virol. 2011, 52, 107–112. [Google Scholar] [CrossRef]
- Levin, M.J.; Bacon, T.H.; Leary, J.J. Resistance of Herpes Simplex Virus Infections to Nucleoside Analogues in HIV-Infected Patients. Clin. Infect. Dis. 2004, 39, S248–S257. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Bestman-Smith, J.; Boivin, G. Resistance of herpesviruses to antiviral drugs: Clinical impacts and molecular mechanisms. Drug Resist. Updat. 2002, 5, 88–114. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; van de Vijver, D.A.; van Loenen, F.B.; Osterhaus, A.D.; Remeijer, L.; Verjans, G.M. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J. Infect. Dis. 2013, 208, 1359–1365. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; de Vries, R.D.; Osterhaus, A.D.M.E.; Remeijer, L.; Verjans, G.M.G.M. Acyclovir-Resistant Corneal HSV-1 Isolates from Patients with Herpetic Keratitis. J. Infect. Dis. 2008, 198, 659–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses 2020, 13, 17. [Google Scholar] [CrossRef]
- Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. [Google Scholar] [CrossRef]
- Taylor, T.J.; Brockman, M.A.; McNamee, E.E.; Knipe, D.M. Herpes simplex virus. Front. Biosci. 2002, 7, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Chamorro, L.; Felip, E.; Ezeonwumelu, I.J.; Margelí, M.; Ballana, E. Cyclin-dependent Kinases as Emerging Targets for Developing Novel Antiviral Therapeutics. Trends Microbiol. 2021, 29, 836–848. [Google Scholar] [CrossRef]
- Schang, L.M.; Phillips, J.; Schaffer, P.A. Requirement for cellular cyclin-dependent kinases in herpes simplex virus replication and transcription. J. Virol. 1998, 72, 5626–5637. [Google Scholar] [CrossRef]
- Schang, L.M.; Rosenberg, A.; Schaffer, P.A. Roscovitine, a Specific Inhibitor of Cellular Cyclin-Dependent Kinases, Inhibits Herpes Simplex Virus DNA Synthesis in the Presence of Viral Early Proteins. J. Virol. 2000, 74, 2107–2120. [Google Scholar] [CrossRef] [Green Version]
- Schang, L.M.; Rosenberg, A.; Schaffer, P.A. Transcription of herpes simplex virus immediate-early and early genes is inhibited by roscovitine, an inhibitor specific for cellular cyclin-dependent kinases. J. Virol. 1999, 73, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Coccaro, E.; Lacasse, J.J. CDK inhibitory nucleoside analogs prevent transcription from viral genomes. Nucleosides Nucleotides Nucleic Acids 2005, 24, 829–837. [Google Scholar] [CrossRef]
- Yamamoto, M.; Onogi, H.; Kii, I.; Yoshida, S.; Iida, K.; Sakai, H.; Abe, M.; Tsubota, T.; Ito, N.; Hosoya, T.; et al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses. J. Clin. Investig. 2014, 124, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Bantly, A.; Schaffer, P.A. Explant-Induced Reactivation of Herpes Simplex Virus Occurs in Neurons Expressing Nuclear cdk2 and cdk4. J. Virol. 2002, 76, 7724–7735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnahan, W.A.; Boldogh, I.; Chi, P.; Thompson, E.; Albrecht, T. Inhibition of Cellular Cdk2 Activity Blocks Human Cytomegalovirus Replication. Virology 1997, 231, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Badia, R.; Angulo, G.; Riveira-Muñoz, E.; Pujantell, M.; Puig, T.; Ramirez, C.; Torres-Torronteras, J.; Martí, R.; Pauls, E.; Clotet, B.; et al. Inhibition of herpes simplex virus type 1 by the CDK6 inhibitor PD-0332991 (palbociclib) through the control of SAMHD1. J. Antimicrob. Chemother. 2015, 71, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.F.; McMichael, M.A.; Leisenfelder, S.A.; Taylor, S.L. Viral and cellular kinases are potential antiviral targets and have a central role in varicella zoster virus pathogenesis. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2004, 1697, 225–231. [Google Scholar] [CrossRef]
- Nelson, P.J.; Gelman, I.H.; Klotman, P.E. Suppression of HIV-1 expression by inhibitors of cyclin-dependent kinases promotes differentiation of infected podocytes. J. Am. Soc. Nephrol. 2001, 12, 2827–2831. [Google Scholar] [CrossRef]
- Guendel, I.; Agbottah, E.T.; Kehn-Hall, K.; Kashanchi, F. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res. Ther. 2010, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Pauls, E.; Badia, R.; Torres-Torronteras, J.; Ruiz, A.; Permanyer, M.; Riveira-Munoz, E.; Clotet, B.; Marti, R.; Ballana, E.; Este, J.A. Palbociclib, a selective inhibitor of cyclin-dependent kinase4/6, blocks HIV-1 reverse transcription through the control of sterile alpha motif and HD domain-containing protein-1 (SAMHD1) activity. Aids 2014, 28, 2213–2222. [Google Scholar] [CrossRef]
- Perwitasari, O.; Yan, X.; O’Donnell, J.; Johnson, S.; Tripp, R.A.; Schor, S.; Einav, S.; Bloom, B.E.; Krouse, A.J.; Gray, L.; et al. Repurposing Kinase Inhibitors as Antiviral Agents to Control Influenza A Virus Replication. ASSAY Drug Dev. Technol. 2015, 13, 638–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Okuyama-Dobashi, K.; Murakami, S.; Chen, W.; Okamoto, T.; Ueda, K.; Hosoya, T.; Matsuura, Y.; Ryo, A.; Tanaka, Y.; et al. Inhibitory effect of CDK9 inhibitor FIT-039 on hepatitis B virus propagation. Antivir. Res. 2016, 133, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, C.; Zhang, J.; Wang, J.; Yu, R.; Wang, D.; Yin, R.; Li, W.; Jiang, T. Guanidine modifications enhance the anti-herpes simplex virus activity of (E,E)-4,6-bis(styryl)-pyrimidine derivatives in vitro and in vivo. Br. J. Pharmacol. 2020, 177, 1568–1588. [Google Scholar] [CrossRef]
- Misra, R.N.; Xiao, H.; Rawlins, D.B.; Shan, W.; Kellar, K.A.; Mulheron, J.G.; Sack, J.S.; Tokarski, J.S.; Kimball, S.D.; Webster, K.R. 1H-Pyrazolo[3,4-b]pyridine inhibitors of cyclin-dependent kinases: Highly potent 2,6-Difluorophenacyl analogues. Bioorganic Med. Chem. Lett. 2003, 13, 2405–2408. [Google Scholar] [CrossRef]
- Mitsuya, H.; Looney, D.J.; Kuno, S.; Ueno, R.; Wong-Staal, F.; Broder, S. Dextran Sulfate Suppression of Viruses in the HIV Family: Inhibition of Virion Binding to CD4+ Cells. Science 1988, 240, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Andreu, S.; von Kobbe, C.; Delgado, P.; Ripa, I.; Buzon, M.J.; Genesca, M.; Girones, N.; Del Moral-Salmoral, J.; Ramirez, G.A.; Zuniga, S.; et al. Dextran sulfate from Leuconostoc mesenteroides B512F exerts potent antiviral activity against SARS-CoV-2 in vitro and in vivo. Front. Microbiol. 2023, 14, 1185504. [Google Scholar] [CrossRef]
- Yamada, H.; Moriishi, E.; Haredy, A.M.; Takenaka, N.; Mori, Y.; Yamanishi, K.; Okamoto, S. Influenza virus neuraminidase contributes to the dextran sulfate-dependent suppressive replication of some influenza A virus strains. Antivir. Res. 2012, 96, 344–352. [Google Scholar] [CrossRef]
- Enserink, J.M.; Kolodner, R.D. An overview of Cdk1-controlled targets and processes. Cell Div. 2010, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Mou, J.; Chen, D.; Deng, Y. Inhibitors of Cyclin-Dependent Kinase 1/2 for Anticancer Treatment. Med. Chem. 2020, 16, 307–325. [Google Scholar] [CrossRef]
- Echalier, A.; Endicott, J.A.; Noble, M.E. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 Knockout Mice Are Viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamle, S.; Ma, B.; He, C.H.; Akosman, B.; Zhou, Y.; Lee, C.-M.; El-Deiry, W.S.; Huntington, K.; Liang, O.; Machan, J.T.; et al. Chitinase 3-like-1 is a therapeutic target that mediates the effects of aging in COVID-19. J. Clin. Investig. 2021, 6, e148749. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Sarver, J.G.; Trabbic, C.J.; Erhardt, P.W.; Schroering, A.; Maltese, W.A. 6-MOMIPP, a novel brain-penetrant anti-mitotic indolyl-chalcone, inhibits glioblastoma growth and viability. Cancer Chemother. Pharmacol. 2018, 83, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, J.F.; Ho, F.; Robertson, E.S.; You, J. Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis. Cancers 2020, 12, 1637. [Google Scholar] [CrossRef]
- De Meo, S.; Dell’oste, V.; Molfetta, R.; Tassinari, V.; Lotti, L.V.; Vespa, S.; Pignoloni, B.; Covino, D.A.; Fantuzzi, L.; Bona, R.; et al. SAMHD1 phosphorylation and cytoplasmic relocalization after human cytomegalovirus infection limits its antiviral activity. PLoS Pathog. 2020, 16, e1008855. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence |
|---|---|---|
| HSV-1 ICP27 | Forward | ATCGCACCTTCTCTGTGGTC |
| Reverse | GCAAATCTTCTGGGGTTTCA | |
| HSV-1 gD | Forward | TACAACCTGACCATCGCTTG |
| Reverse | GCCCCCAGAGACTTGTTGTA | |
| HSV-1 ICP8 | Forward | GTCGTTACCGAGGGCTTCAA |
| Reverse | GTTACCTTGTCCGAGCCTCC | |
| β-actin | Forward | CTCCATCCTGGCCTCGCTGT |
| Reverse | GCTGTCACCTTCACCGTTCC |
| CDK Inhibitor | Target(s) | CC50 (µM) | IC50 (µM) | Selective Index |
|---|---|---|---|---|
| Abemaciclib | CDK4/6 | 29.11 | ND | - |
| BGG463 | CDK2 | 18.46 | ND | - |
| BMS-265246 | CDK1/2 | >500 | 0.08 | >6250 |
| Butyrolactone I | CDK1 | 151.20 | ND | - |
| CVT-313 | CDK2 | 6.38 | ND | - |
| Dinaciclib | CDK1/2/5/9 | 1.13 | ND | - |
| Flavopiridol | CDK1/2/4 | 0.16 | ND | - |
| LDC000067 | CDK9 | 14.37 | ND | - |
| MSC2530818 | CDK8 | 138.50 | 25 | 5.54 |
| NU6300 | CDK2 | 13.83 | ND | - |
| PNU112455A | CDK2/5 | 156.40 | ND | - |
| Purvalanol B | CDK2/5 | 73.52 | ND | - |
| Ro-3306 | CDK1/2 | 14.11 | ND | - |
| Seliciclib | CDK2/5/7 | 18.47 | ND | - |
| THZ2 | CDK7 | 0.42 | ND | - |
| Acyclovir (Reference) | Herpesvirus POL | >100 | 0.42 | >238.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Yu, Y.; Li, Z.; Gao, Y.; Zhang, H.; Zhang, M.; Cao, W.; Peng, Q.; Chen, X. BMS-265246, a Cyclin-Dependent Kinase Inhibitor, Inhibits the Infection of Herpes Simplex Virus Type 1. Viruses 2023, 15, 1642. https://doi.org/10.3390/v15081642
Jiang L, Yu Y, Li Z, Gao Y, Zhang H, Zhang M, Cao W, Peng Q, Chen X. BMS-265246, a Cyclin-Dependent Kinase Inhibitor, Inhibits the Infection of Herpes Simplex Virus Type 1. Viruses. 2023; 15(8):1642. https://doi.org/10.3390/v15081642
Chicago/Turabian StyleJiang, Lefang, Yang Yu, Zhuogang Li, Yarou Gao, Haonan Zhang, Mingxin Zhang, Weihua Cao, Qun Peng, and Xulin Chen. 2023. "BMS-265246, a Cyclin-Dependent Kinase Inhibitor, Inhibits the Infection of Herpes Simplex Virus Type 1" Viruses 15, no. 8: 1642. https://doi.org/10.3390/v15081642
APA StyleJiang, L., Yu, Y., Li, Z., Gao, Y., Zhang, H., Zhang, M., Cao, W., Peng, Q., & Chen, X. (2023). BMS-265246, a Cyclin-Dependent Kinase Inhibitor, Inhibits the Infection of Herpes Simplex Virus Type 1. Viruses, 15(8), 1642. https://doi.org/10.3390/v15081642