
Genetic Diversity of Type A Influenza Viruses Found in Swine Herds in Northwestern Poland from 2017 to 2019: The One Health Perspective

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Samples from Swine and Isolation of Viral RNA
2.2. Confirmation of Influenza A Virus Infection in Collected Samples
2.3. Virus Isolation from Madin-Darby Canine Kidney (MDCK) Cell Culture
2.4. Nucleic Acid Extraction from Viral Isolates and Amplification of the Whole Influenza A Virus Genome Segments
2.5. Preparation of Sequencing Library
2.6. Whole-Genome Sequencing Using MiniSeq Platform (Illumina), Followed by Bioinformatical and Phylogenetic Analysis of Gathered Data
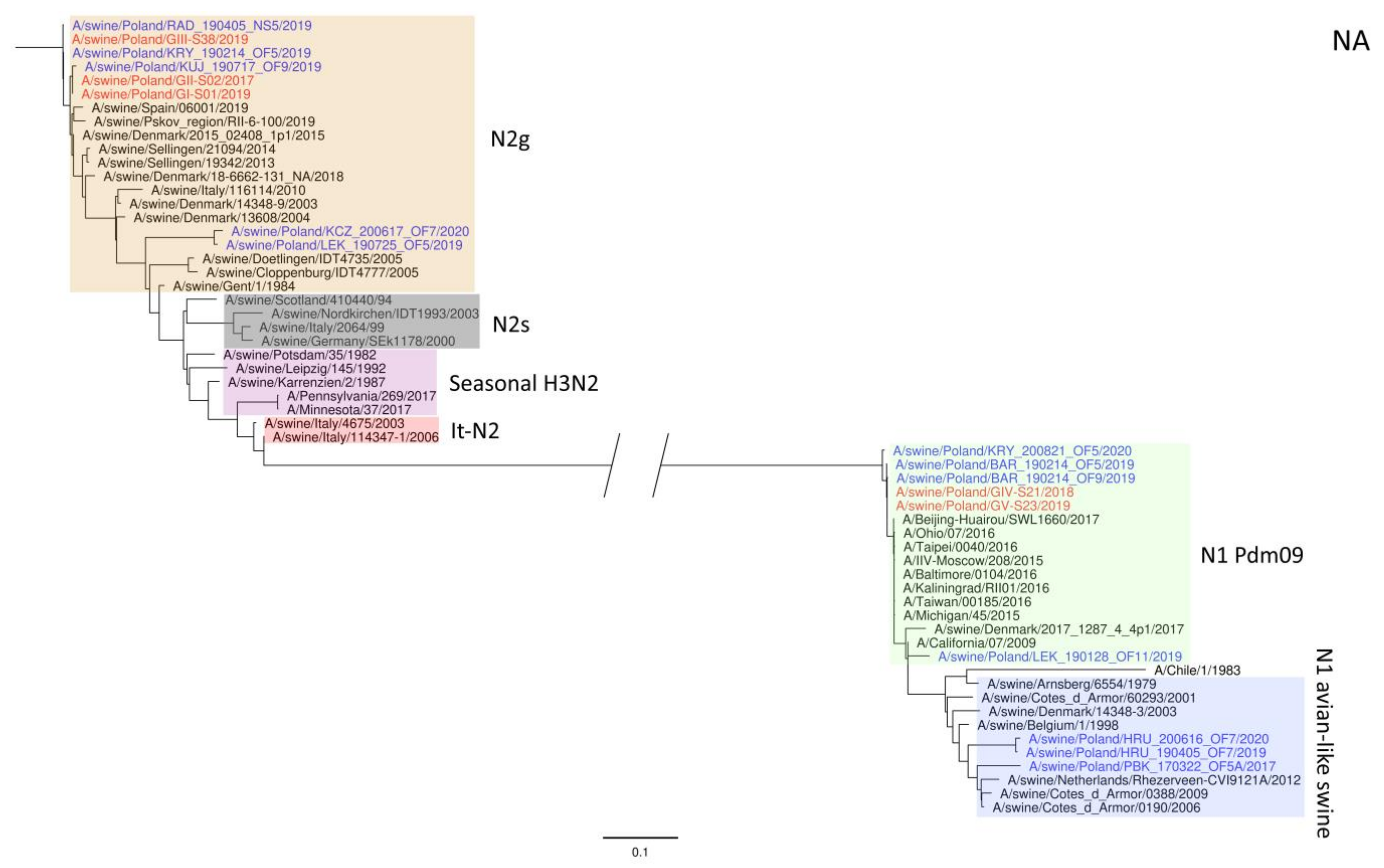
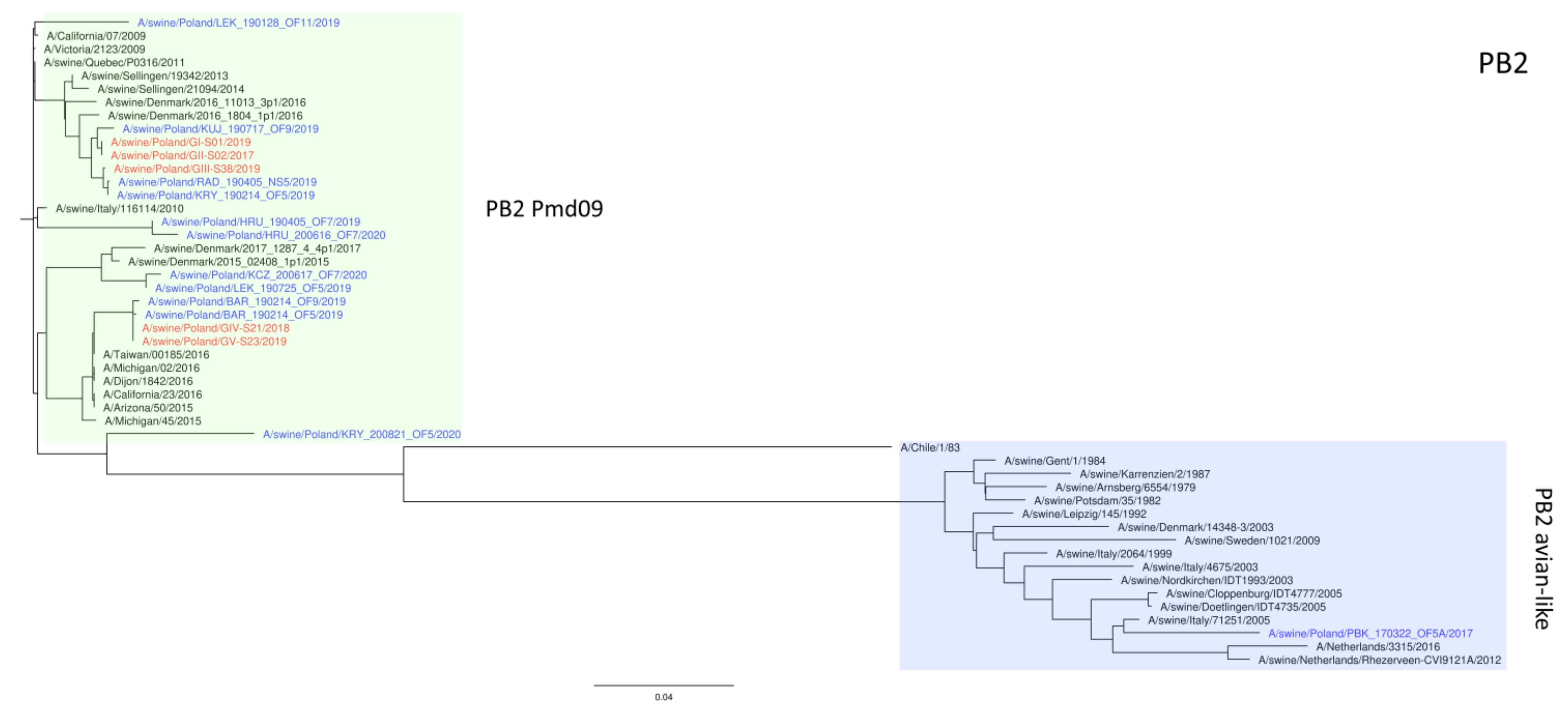
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, S.-W.; Webby, R.J.; Webster, R.G. Evolution and Ecology of Influenza A Viruses. In Influenza Pathogenesis and Control—Volume I; Compans, R.W., Oldstone, M.B.A., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2014; pp. 359–375. ISBN 978-3-319-11155-1. [Google Scholar]
- Suarez, D.L. Influenza A Virus. In Animal Influenza; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 1–30. ISBN 978-1-118-92434-1. [Google Scholar]
- Goneau, L.W.; Mehta, K.; Wong, J.; L’Huillier, A.G.; Gubbay, J.B. Zoonotic Influenza and Human Health—Part 1: Virology and Epidemiology of Zoonotic Influenzas. Curr. Infect. Dis. Rep. 2018, 20, 37. [Google Scholar] [CrossRef] [PubMed]
- Mänz, B.; Schwemmle, M.; Brunotte, L. Adaptation of Avian Influenza A Virus Polymerase in Mammals To Overcome the Host Species Barrier. J. Virol. 2013, 87, 7200–7209. [Google Scholar] [CrossRef] [PubMed]
- Linster, M.; van Boheemen, S.; de Graaf, M.; Schrauwen, E.J.A.; Lexmond, P.; Mänz, B.; Bestebroer, T.M.; Baumann, J.; van Riel, D.; Rimmelzwaan, G.F.; et al. Identification, Characterization, and Natural Selection of Mutations Driving Airborne Transmission of A/H5N1 Virus. Cell 2014, 157, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza Viruses and MRNA Splicing: Doing More with Less. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Reperant, L.A.; Grenfell, B.T.; Osterhaus, A.D.M.E. Quantifying the Risk of Pandemic Influenza Virus Evolution by Mutation and Re-Assortment. Vaccine 2015, 33, 6955–6966. [Google Scholar] [CrossRef] [PubMed]
- Feldblyum, T.V.; Segal, D.M. Seasonal and Pandemic Influenza Surveillance and Disease Severity. Glob. Virol. I—Identifying Investig. Viral Dis. 2015, 761–789. [Google Scholar] [CrossRef]
- Petrova, V.N. The Evolution of Seasonal Influenza Viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Forrest, H.L.; Webster, R.G. Perspectives on Influenza Evolution and the Role of Research. Anim. Health Res. Rev. 2010, 11, 3–18. [Google Scholar] [CrossRef]
- Scholtissek, C. Pigs as ‘Mixing Vessels’ for the Creation of New Pandemic Influenza A Viruses. Med. Princ. Pract. 1990, 2, 65–71. [Google Scholar] [CrossRef]
- Krumbholz, A.; Lange, J.; Sauerbrei, A.; Groth, M.; Platzer, M.; Kanrai, P.; Pleschka, S.; Scholtissek, C.; Büttner, M.; Dürrwald, R.; et al. Origin of the European Avian-like Swine Influenza Viruses. J. Gen. Virol. 2014, 95, 2372–2376. [Google Scholar] [CrossRef]
- Campitelli, L.; Donatelli, I.; Foni, E.; Castrucci, M.R.; Fabiani, C.; Kawaoka, Y.; Krauss, S.; Webster, R.G. Continued Evolution of H1N1 and H3N2 Influenza Viruses in Pigs in Italy. Virology 1997, 232, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.H.; Harris, P.A.; McCauley, J.W.; Alexander, D.J. Multiple Genetic Reassortment of Avian and Human Influenza A Viruses in European Pigs, Resulting in the Emergence of an H1N2 Virus of Novel Genotype. J. Gen. Virol. 1998, 79 Pt 12, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Mena, I.; Nelson, M.I.; Quezada-Monroy, F.; Dutta, J.; Cortes-Fernández, R.; Lara-Puente, J.H.; Castro-Peralta, F.; Cunha, L.F.; Trovão, N.S.; Lozano-Dubernard, B.; et al. Origins of the 2009 H1N1 Influenza Pandemic in Swine in Mexico. eLife 2016, 5, e16777. [Google Scholar] [CrossRef]
- Trebbien, R.; Bragstad, K.; Larsen, L.E.; Nielsen, J.; Bøtner, A.; Heegaard, P.M.H.; Fomsgaard, A.; Viuff, B.; Hjulsager, C.K. Genetic and Biological Characterisation of an Avian-like H1N2 Swine Influenza Virus Generated by Reassortment of Circulating Avian-Like H1N1 and H3N2 Subtypes in Denmark. Virol. J. 2013, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Krog, J.S.; Hjulsager, C.K.; Larsen, M.A.; Larsen, L.E. Triple-reassortant Influenza A Virus with H3 of Human Seasonal Origin, NA of Swine Origin, and Internal A(H1N1) Pandemic 2009 Genes Is Established in Danish Pigs. Influenza Other Respir Viruses 2017, 11, 298–303. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Krumbholz, A.; Lange, J.; Philipps, A.; Dürrwald, R. Displacement of the Gent/1999 Human-like Swine H1N2 Influenza A Virus Lineage by Novel H1N2 Reassortants in Germany. Arch. Virol. 2020, 165, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Chiapponi, C.; Prosperi, A.; Moreno, A.; Baioni, L.; Faccini, S.; Manfredi, R.; Zanni, I.; Gabbi, V.; Calanchi, I.; Fusaro, A.; et al. Genetic Variability among Swine Influenza Viruses in Italy: Data Analysis of the Period 2017–2020. Viruses 2021, 14, 47. [Google Scholar] [CrossRef]
- Chepkwony, S.; Parys, A.; Vandoorn, E.; Stadejek, W.; Xie, J.; King, J.; Graaf, A.; Pohlmann, A.; Beer, M.; Harder, T.; et al. Genetic and Antigenic Evolution of H1 Swine Influenza A Viruses Isolated in Belgium and the Netherlands from 2014 through 2019. Sci. Rep. 2021, 11, 11276. [Google Scholar] [CrossRef]
- Chastagner, A.; Hervé, S.; Bonin, E.; Quéguiner, S.; Hirchaud, E.; Henritzi, D.; Béven, V.; Gorin, S.; Barbier, N.; Blanchard, Y.; et al. Spatiotemporal Distribution and Evolution of the A/H1N1 2009 Pandemic Influenza Virus in Pigs in France from 2009 to 2017: Identification of a Potential Swine-Specific Lineage. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Watson, S.J.; Langat, P.; Reid, S.M.; Lam, T.T.-Y.; Cotten, M.; Kelly, M.; Van Reeth, K.; Qiu, Y.; Simon, G.; Bonin, E.; et al. Molecular Epidemiology and Evolution of Influenza Viruses Circulating within European Swine between 2009 and 2013. J. Virol. 2015, 89, 9920–9931. [Google Scholar] [CrossRef]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627.e6. [Google Scholar] [CrossRef]
- Anderson, T.K.; Macken, C.A.; Lewis, N.S.; Scheuermann, R.H.; Van Reeth, K.; Brown, I.H.; Swenson, S.L.; Simon, G.; Saito, T.; Berhane, Y.; et al. A Phylogeny-Based Global Nomenclature System and Automated Annotation Tool for H1 Hemagglutinin Genes from Swine Influenza A Viruses. mSphere 2016, 1, e00275-16. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Scholtissek, C.; Ludwig, S. Genetics, Evolution, and the Zoonotic Capacity of European Swine Influenza Viruses. Curr. Top. Microbiol. Immunol. 2013, 370, 29–55. [Google Scholar] [CrossRef] [PubMed]
- Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. Emergence of a Novel Swine-Origin Influenza A (H1N1) Virus in Humans. N. Engl. J. Med. 2009, 360, 2605–2615. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xiao, Y.; Liu, J.; Wang, D.; Li, F.; Wang, C.; Li, C.; Zhu, J.; Song, J.; Sun, H.; et al. Prevalent Eurasian Avian-like H1N1 Swine Influenza Virus with 2009 Pandemic Viral Genes Facilitating Human Infection. Porc. Natl. Acad. Sci. USA 2020, 117, 17204–17210. [Google Scholar] [CrossRef] [PubMed]
- Er, J.C.; Lium, B.; Framstad, T. Antibodies of Influenza A(H1N1)Pdm09 Virus in Pigs’ Sera Cross-React with Other Influenza A Virus Subtypes. A Retrospective Epidemiological Interpretation of Norway’s Serosurveillance Data from 2009–2017. Epidemiol. Infect. 2020, 148, e73. [Google Scholar] [CrossRef] [PubMed]
- Czyżewska-Dors, E.; Dors, A.; Kwit, K.; Pejsak, Z.; Pomorska-Mól, M. Serological Survey of the Influenza a Virus in Polish Farrow-to-Finish Pig Herds in 2011–2015. J. Vet. Res. 2017, 61, 157–161. [Google Scholar] [CrossRef]
- Zoonotic Influenza- Annual Epidemiological Report for 2014. Available online: https://www.ecdc.europa.eu/en/publications-data/zoonotic-influenza-annual-epidemiological-report-2014 (accessed on 18 February 2022).
- Threat Assessment Brief: Eurasian Avian-like A(H1N1) Swine Influenza Viruses. Available online: https://www.ecdc.europa.eu/en/publications-data/threat-assessment-brief-eurasian-avian-ah1n1-swine-influenza-viruses (accessed on 18 February 2022).
- Jhung, M.A.; Epperson, S.; Biggerstaff, M.; Allen, D.; Balish, A.; Barnes, N.; Beaudoin, A.; Berman, L.; Bidol, S.; Blanton, L.; et al. Outbreak of Variant Influenza A(H3N2) Virus in the United States. Clin. Infect. Dis. 2013, 57, 1703–1712. [Google Scholar] [CrossRef]
- Nelson, M.I.; Vincent, A.L. Reverse Zoonosis of Influenza to Swine: New Perspectives on the Human-Animal Interface. Trends Microbiol. 2015, 23, 142–153. [Google Scholar] [CrossRef]
- Krammer, F.; Palese, P. Advances in the Development of Influenza Virus Vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef]
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- Soh, Y.S.; Moncla, L.H.; Eguia, R.; Bedford, T.; Bloom, J.D. Comprehensive Mapping of Adaptation of the Avian Influenza Polymerase Protein PB2 to Humans. eLife 2019, 8, e45079. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Gambhir, M.; Finelli, L.; Swerdlow, D.L.; Ostroff, S.; Reed, C. Transmissibility of Variant Influenza From Swine to Humans: A Modeling Approach. Clin. Infect. Dis. 2013, 57, S16–S22. [Google Scholar] [CrossRef]
- Cauchemez, S.; Epperson, S.; Biggerstaff, M.; Swerdlow, D.; Finelli, L.; Ferguson, N.M. Using Routine Surveillance Data to Estimate the Epidemic Potential of Emerging Zoonoses: Application to the Emergence of US Swine Origin Influenza A H3N2v Virus. PLoS Med. 2013, 10, e1001399. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Lowen, A.C. Implications of Segment Mismatch for Influenza A Virus Evolution. J. Gen. Virol. 2018, 99, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Chit, A.; Soobiah, C.; Hallett, D.; Meier, G.; Chen, M.H.; Tashkandi, M.; Bauch, C.T.; Loeb, M. Comparing Influenza Vaccine Efficacy against Mismatched and Matched Strains: A Systematic Review and Meta-Analysis. BMC Med. 2013, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.F.; Neumann, G.; Kawaoka, Y. Orthomyxoviruses. In Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 1186–1241. [Google Scholar]
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X.; et al. The Genesis and Source of the H7N9 Influenza Viruses Causing Human Infections in China. Nature 2013, 502, 241–244. [Google Scholar] [CrossRef]
- Smith, G.J.D.; Bahl, J.; Vijaykrishna, D.; Zhang, J.; Poon, L.L.M.; Chen, H.; Webster, R.G.; Peiris, J.S.M.; Guan, Y. Dating the Emergence of Pandemic Influenza Viruses. Porc. Natl. Acad. Sci. USA 2009, 106, 11709–11712. [Google Scholar] [CrossRef]
- Guan, Y.; Shortridge, K.F.; Krauss, S.; Webster, R.G. Molecular Characterization of H9N2 Influenza Viruses: Were They the Donors of the “Internal” Genes of H5N1 Viruses in Hong Kong? Proc. Natl. Acad. Sci. USA 1999, 96, 9363–9367. [Google Scholar] [CrossRef]
- Nelson, M.I.; Gramer, M.R.; Vincent, A.L.; Holmes, E.C. Global Transmission of Influenza Viruses from Humans to Swine. J. Gen. Virol. 2012, 93, 2195–2203. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Forgie, S.E.; Keenliside, J.; Wilkinson, C.; Webby, R.; Lu, P.; Sorensen, O.; Fonseca, K.; Barman, S.; Rubrum, A.; Stigger, E.; et al. Swine Outbreak of Pandemic Influenza A Virus on a Canadian Research Farm Supports Human-to-Swine Transmission. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2011, 52, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.; Larsen, L.E.; Dürrwald, R.; Foni, E.; Harder, T.; Van Reeth, K.; Markowska-Daniel, I.; Reid, S.M.; Dan, A.; Maldonado, J.; et al. European Surveillance Network for Influenza in Pigs: Surveillance Programs, Diagnostic Tools and Swine Influenza Virus Subtypes Identified in 14 European Countries from 2010 to 2013. PLoS ONE 2014, 9, e115815. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.H. History and Epidemiology of Swine Influenza in Europe. In Swine Influenza; Richt, J.A., Webby, R.J., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 133–146. ISBN 978-3-642-36871-4. [Google Scholar]
- Hofshagen, M.; Gjerset, B.; Er, C.; Tarpai, A.; Brun, E.; Dannevig, B.; Bruheim, T.; Fostad, I.G.; Iversen, B.; Hungnes, O.; et al. Pandemic Influenza A(H1N1)v: Human to Pig Transmission in Norway? Eurosurveillance 2009, 14, 19406. [Google Scholar] [CrossRef] [PubMed]
- Grøntvedt, C.A.; Er, C.; Gjerset, B.; Germundsson, A.; Framstad, T.; Brun, E.; Jørgensen, A.; Lium, B. Clinical Impact of Infection with Pandemic Influenza (H1N1) 2009 Virus in Naïve Nucleus and Multiplier Pig Herds in Norway. Influenza Res. Treat. 2011, 2011, 163745. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Hause, B.; Stigger-Rosser, E.; Darnell, D.; Corzo, C.; Juleen, K.; Simonson, R.; Brockwell-Staats, C.; Rubrum, A.; Wang, D.; et al. Multiple Reassortment between Pandemic (H1N1) 2009 and Endemic Influenza Viruses in Pigs, United States. Emerg. Infect. Dis. 2011, 17, 1624–1629. [Google Scholar] [CrossRef]
- Rajão, D.S.; Walia, R.R.; Campbell, B.; Gauger, P.C.; Janas-Martindale, A.; Killian, M.L.; Vincent, A.L. Reassortment between Swine H3N2 and 2009 Pandemic H1N1 in the United States Resulted in Influenza A Viruses with Diverse Genetic Constellations with Variable Virulence in Pigs. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Rovida, F.; Piralla, A.; Marzani, F.C.; Moreno, A.; Campanini, G.; Mojoli, F.; Pozzi, M.; Girello, A.; Chiapponi, C.; Vezzoli, F.; et al. Swine influenza A (H1N1) Virus (SIV) Infection Requiring Extracorporeal Life Support in an Immunocompetent Adult Patient with Indirect Exposure to Pigs, Italy, October 2016. Eurosurveillance 2017, 22, 30456. [Google Scholar] [CrossRef]
- Li, X.; Guo, L.; Liu, C.; Cheng, Y.; Kong, M.; Yang, L.; Zhuang, Z.; Liu, J.; Zou, M.; Dong, X.; et al. Human Infection with a Novel Reassortant Eurasian-Avian Lineage Swine H1N1 Virus in Northern China. Emerg. Microbes Infect. 2019, 8, 1535–1545. [Google Scholar] [CrossRef]
- Fraaij, P.L.A.; Wildschut, E.D.; Houmes, R.J.; Swaan, C.M.; Hoebe, C.J.; de Jonge, H.C.C.; Tolsma, P.; de Kleer, I.; Pas, S.D.; Oude Munnink, B.B.; et al. Severe Acute Respiratory Infection Caused by Swine Influenza Virus in a Child Necessitating Extracorporeal Membrane Oxygenation (ECMO), the Netherlands, October 2016. Eurosurveillance 2016, 21, 30416. [Google Scholar] [CrossRef]
- Adlhoch, C.; Penttinen, P. Letter to the Editor: Just a Coincidence? Two Severe Human Cases Due to Swine Influenza (SIV) A(H1N1)v in Europe, October 2016. Eurosurveillance 2017, 22, 30478. [Google Scholar] [CrossRef]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic Potential of Influenza A Viruses: A Comprehensive Overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Webby, R.; Lam, T.T.Y.; Smith, D.K.; Peiris, J.S.M.; Guan, Y. History of Swine Influenza Viruses in Asia. Curr. Top. Microbiol. Immunol. 2013, 370, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, A.; Vincent, A.L.; Gramer, M.E.; Lager, K.M.; Ciacci-Zanella, J.R. Contemporary Epidemiology of North American Lineage Triple Reassortant Influenza A Viruses in Pigs. Curr. Top. Microbiol. Immunol. 2013, 370, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Gabanelli, E.; Sozzi, E.; Lelli, D.; Chiapponi, C.; Ciccozzi, M.; Zehender, G.; Cordioli, P. Different Evolutionary Trends of Swine H1N2 Influenza Viruses in Italy Compared to European Viruses. Vet. Res. 2013, 44, 112. [Google Scholar] [CrossRef]
- Lepek, K.; Pajak, B.; Rabalski, L.; Urbaniak, K.; Kucharczyk, K.; Markowska-Daniel, I.; Szewczyk, B. Analysis of Coinfections with A/H1N1 Strain Variants among Pigs in Poland by Multitemperature Single-Strand Conformational Polymorphism. BioMed. Res. Int. 2015, 2015, e535908. [Google Scholar] [CrossRef]
- Hu, J.; Hu, Z.; Wei, Y.; Zhang, M.; Wang, S.; Tong, Q.; Sun, H.; Pu, J.; Liu, J.; Sun, Y. Mutations in PB2 and HA Are Crucial for the Increased Virulence and Transmissibility of H1N1 Swine Influenza Virus in Mammalian Models. Vet. Microbiol. 2022, 265, 109314. [Google Scholar] [CrossRef]
- Yamaji, H. Suitability and Perspectives on Using Recombinant Insect Cells for the Production of Virus-like Particles. Appl. Microbiol. Biotechnol. 2014, 98, 1963–1970. [Google Scholar] [CrossRef]
- Czudai-Matwich, V.; Otte, A.; Matrosovich, M.; Gabriel, G.; Klenk, H.-D. PB2 Mutations D701N and S714R Promote Adaptation of an Influenza H5N1 Virus to a Mammalian Host. J. Virol. 2014, 88, 8735–8742. [Google Scholar] [CrossRef]
- Yamada, S.; Hatta, M.; Staker, B.L.; Watanabe, S.; Imai, M.; Shinya, K.; Sakai-Tagawa, Y.; Ito, M.; Ozawa, M.; Watanabe, T.; et al. Biological and Structural Characterization of a Host-Adapting Amino Acid in Influenza Virus. PLoS Pathog. 2010, 6, e1001034. [Google Scholar] [CrossRef]
- Thompson, A.J.; Paulson, J.C. Adaptation of Influenza Viruses to Human Airway Receptors. J. Biol. Chem. 2021, 296, 100017. [Google Scholar] [CrossRef] [PubMed]
- Hennig, C.; Graaf, A.; Petric, P.P.; Graf, L.; Schwemmle, M.; Beer, M.; Harder, T. Are Pigs Overestimated as a Source of Zoonotic Influenza Viruses? Porc. Health Manag. 2022, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Wedde, M.; Wählisch, S.; Wolff, T.; Schweiger, B. Predominance of HA-222D/G Polymorphism in Influenza A(H1N1)Pdm09 Viruses Associated with Fatal and Severe Outcomes Recently Circulating in Germany. PLoS ONE 2013, 8, e57059. [Google Scholar] [CrossRef]
- Zhu, X.; McBride, R.; Nycholat, C.M.; Yu, W.; Paulson, J.C.; Wilson, I.A. Influenza Virus Neuraminidases with Reduced Enzymatic Activity That Avidly Bind Sialic Acid Receptors. J. Virol. 2012, 86, 13371–13383. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.P.; Gregory, V.; Collins, P.; Kloess, J.; Wharton, S.; Cattle, N.; Lackenby, A.; Daniels, R.; Hay, A. Neuraminidase Receptor Binding Variants of Human Influenza A(H3N2) Viruses Resulting from Substitution of Aspartic Acid 151 in the Catalytic Site: A Role in Virus Attachment? J. Virol. 2010, 84, 6769–6781. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, N.; Woźniak, A.; Pauwels, M.; Coppens, S.; Nauwynck, H.; Cybulski, P.; Theuns, S.; Stadejek, T. Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (SwIAV) Directly from Oral Fluids Collected in Polish Pig Herds. Viruses 2023, 15, 435. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Localization | Time of Collection | Number of Samples | Number of Confirmed Influenza Infections | Subtype | HA Clade | NA Lineage |
|---|---|---|---|---|---|---|
| 01KO | 05/2017 | 35 | 0 | - | - | - |
| 02SY | 07/2017 | 45 | 24 (53%) | H1N2 | 1C.2 | N2g |
| 03KA | 05/2018 | 30 | 16 (53%) | n/d | n/d | n/d |
| 04BA | 10/2018 | 40 | 19 (47.5%) | H1N1 | 1A.3.3.2 | pdm |
| 05PA | 11/2018 | 20 | 0 | - | - | - |
| 06KU | 03/2019 | 40 | 22 (55%) | H1N2 | 1C.2 | N2g |
| 07RA | 03/2019 | 20 | 16 (80%) | H1N2 | 1C.2 | N2g |
| 08KO | 06/2019 | 21 | 0 | - | - | - |
| 04BA | 07/2019 | 35 | 15 (42.8%) | H1N1 | 1A.3.3.2 | pdm |
| 09WI | 09/2019 | 25 | 0 | - | - | - |
| 10MI | 10/2019 | 30 | 0 | - | - | - |
| 11GI | 10/2019 | 35 | 0 | - | - | - |
| TOTAL | 376 | 112 (29.8%) |
| Genetic Variant | Corresponding Strain | Accession Number |
|---|---|---|
| G I | A/swine/Poland/GI-S01/2019(H1N2) | EPI_ISL_17952486 |
| G II | A/swine/Poland/GII-S02/2017(H1N2) | EPI_ISL_17952487 |
| G III | A/swine/Poland/GIII-S38/2019(H1N2) | EPI_ISL_17952488 |
| G IV | A/swine/Poland/GIV-S21/2018(H1N1) | EPI_ISL_17952489 |
| G V | A/swine/Poland/GV-S23/2019(H1N1) | EPI_ISL_17952490 |
| Subtype | Genotype | HA | NA | PB2 | PB1 | PA | NP | M | NS | % of Analyzed Samples |
|---|---|---|---|---|---|---|---|---|---|---|
| H1N1 | P | 1A.3.3.2 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | 36.6% |
| H1N2 | T | 1C.2 | N2g | pdm | pdm | pdm | pdm | pdm | pdm | 63.4% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabalski, L.; Kosinski, M.; Cybulski, P.; Stadejek, T.; Lepek, K. Genetic Diversity of Type A Influenza Viruses Found in Swine Herds in Northwestern Poland from 2017 to 2019: The One Health Perspective. Viruses 2023, 15, 1893. https://doi.org/10.3390/v15091893
Rabalski L, Kosinski M, Cybulski P, Stadejek T, Lepek K. Genetic Diversity of Type A Influenza Viruses Found in Swine Herds in Northwestern Poland from 2017 to 2019: The One Health Perspective. Viruses. 2023; 15(9):1893. https://doi.org/10.3390/v15091893
Chicago/Turabian StyleRabalski, Lukasz, Maciej Kosinski, Piotr Cybulski, Tomasz Stadejek, and Krzysztof Lepek. 2023. "Genetic Diversity of Type A Influenza Viruses Found in Swine Herds in Northwestern Poland from 2017 to 2019: The One Health Perspective" Viruses 15, no. 9: 1893. https://doi.org/10.3390/v15091893
APA StyleRabalski, L., Kosinski, M., Cybulski, P., Stadejek, T., & Lepek, K. (2023). Genetic Diversity of Type A Influenza Viruses Found in Swine Herds in Northwestern Poland from 2017 to 2019: The One Health Perspective. Viruses, 15(9), 1893. https://doi.org/10.3390/v15091893