
Retinoic Acid-Mediated Inhibition of Mouse Coronavirus Replication Is Dependent on IRF3 and CaMKK

 , ,
, ,  ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultured Cells and Virus
2.2. Reagents
2.3. RA Cell Survival Quantification
2.4. MHV Infection
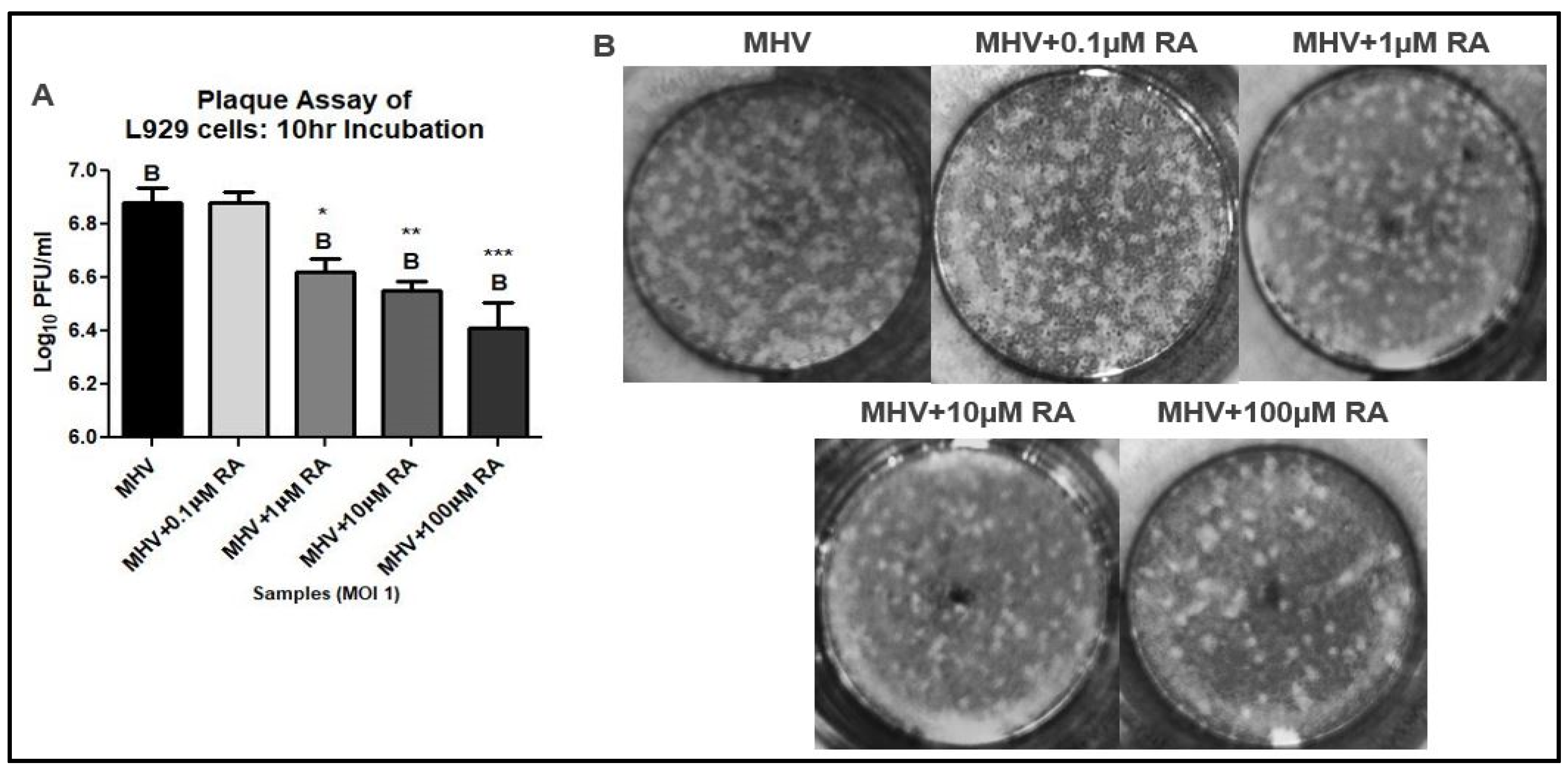
2.5. Viral Plaque Assay
2.6. RNA Extraction and qRT-PCR
2.7. Protein Extraction and Western Blot Analysis
2.8. IFN Regulatory Factor Activation Assay
2.9. RNA Extraction and mRNA-Sequencing Analysis
2.10. Differential Gene Expression Analysis
2.11. Heatmap and Volcano Plot Generation
2.12. Statistical Analysis
3. Results
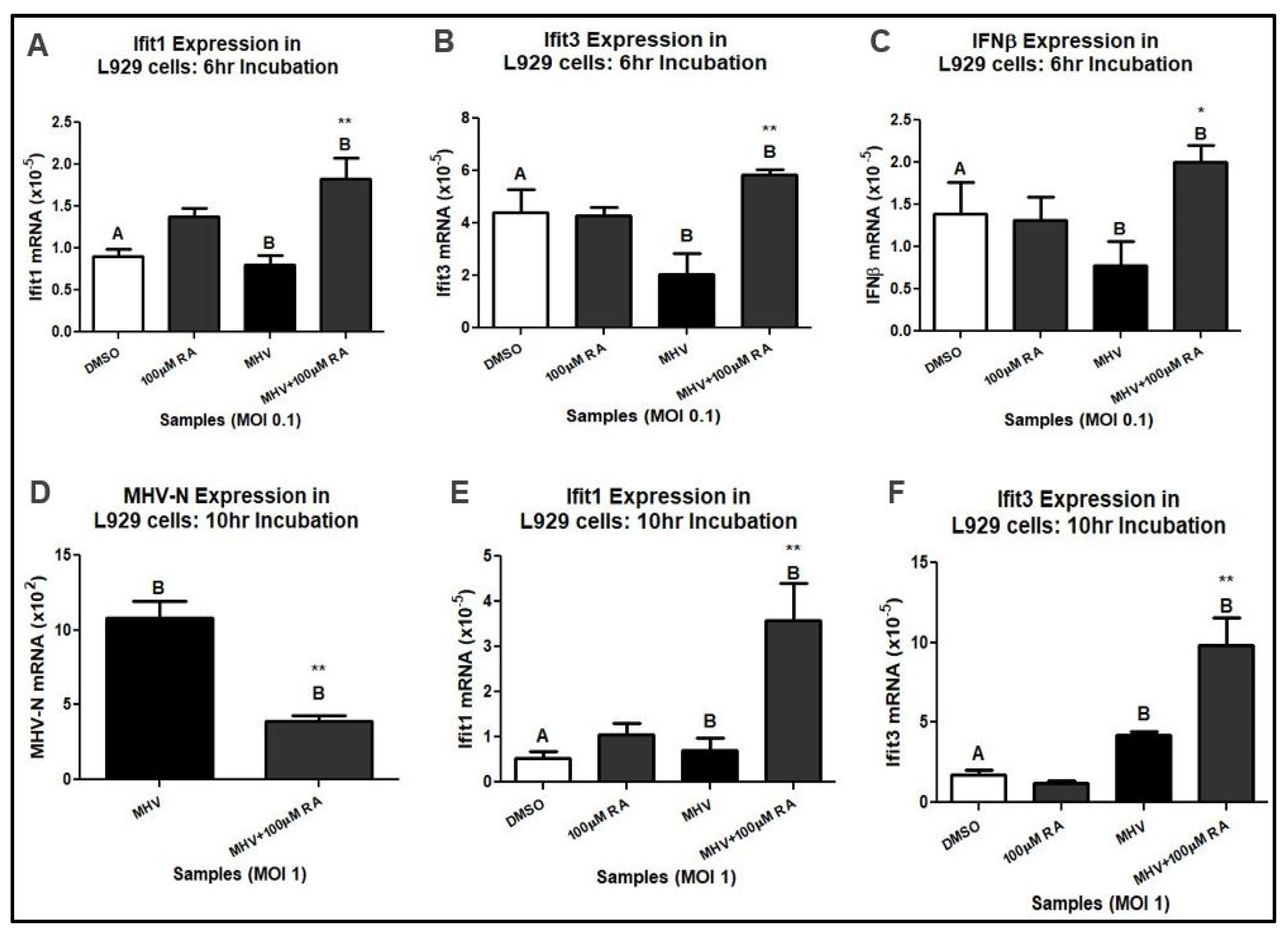
3.1. RA Confers Protection against MHV Infection
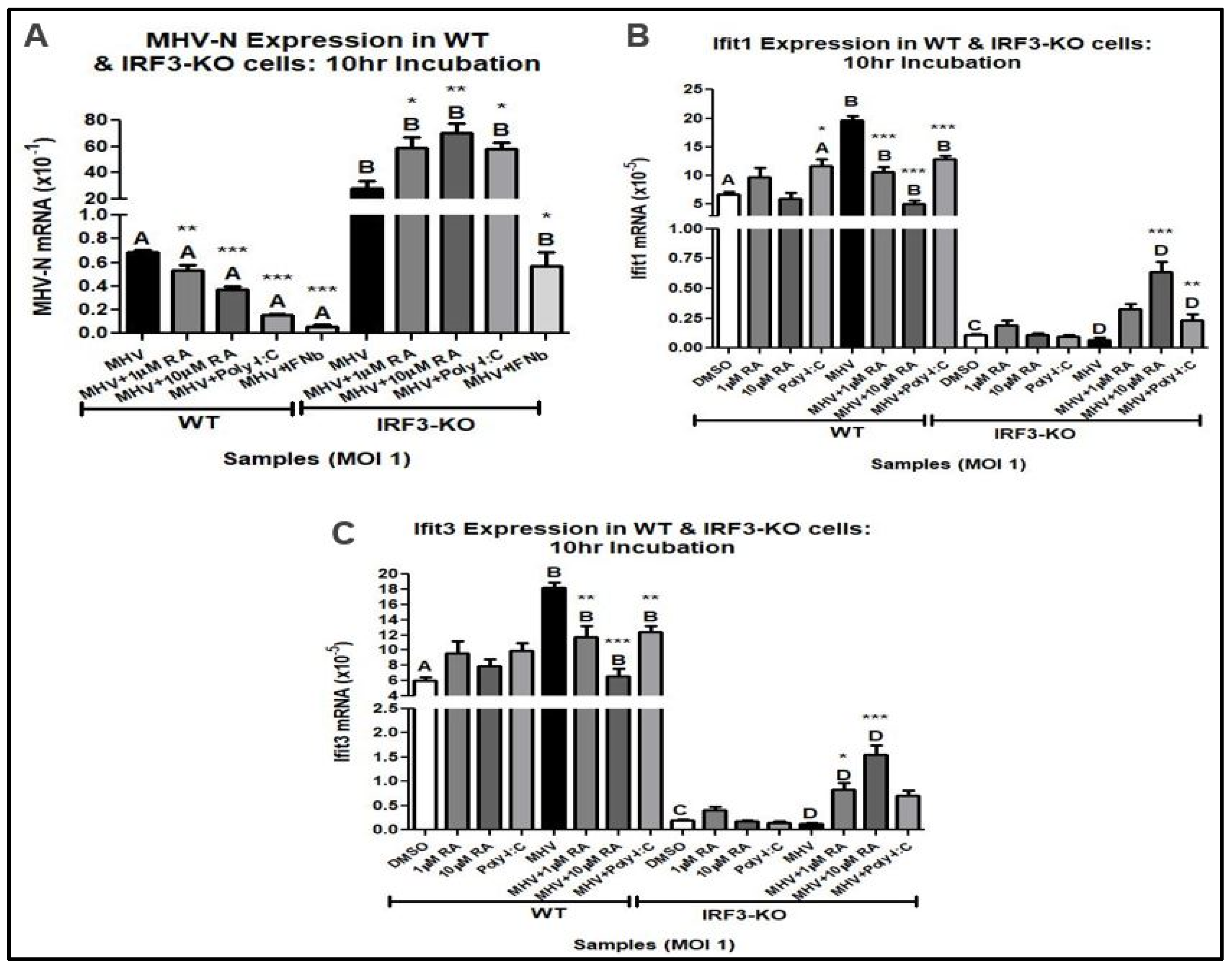
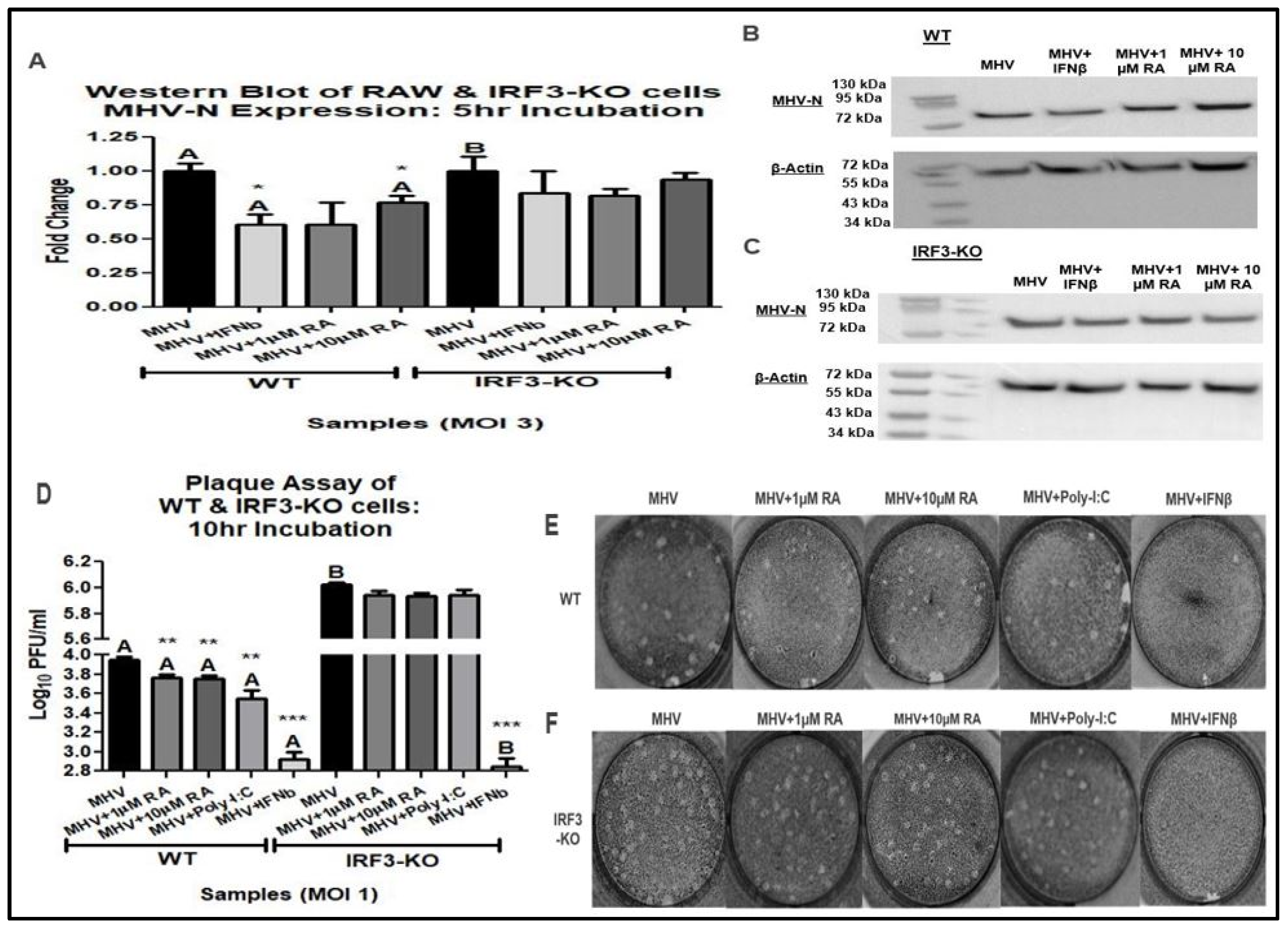
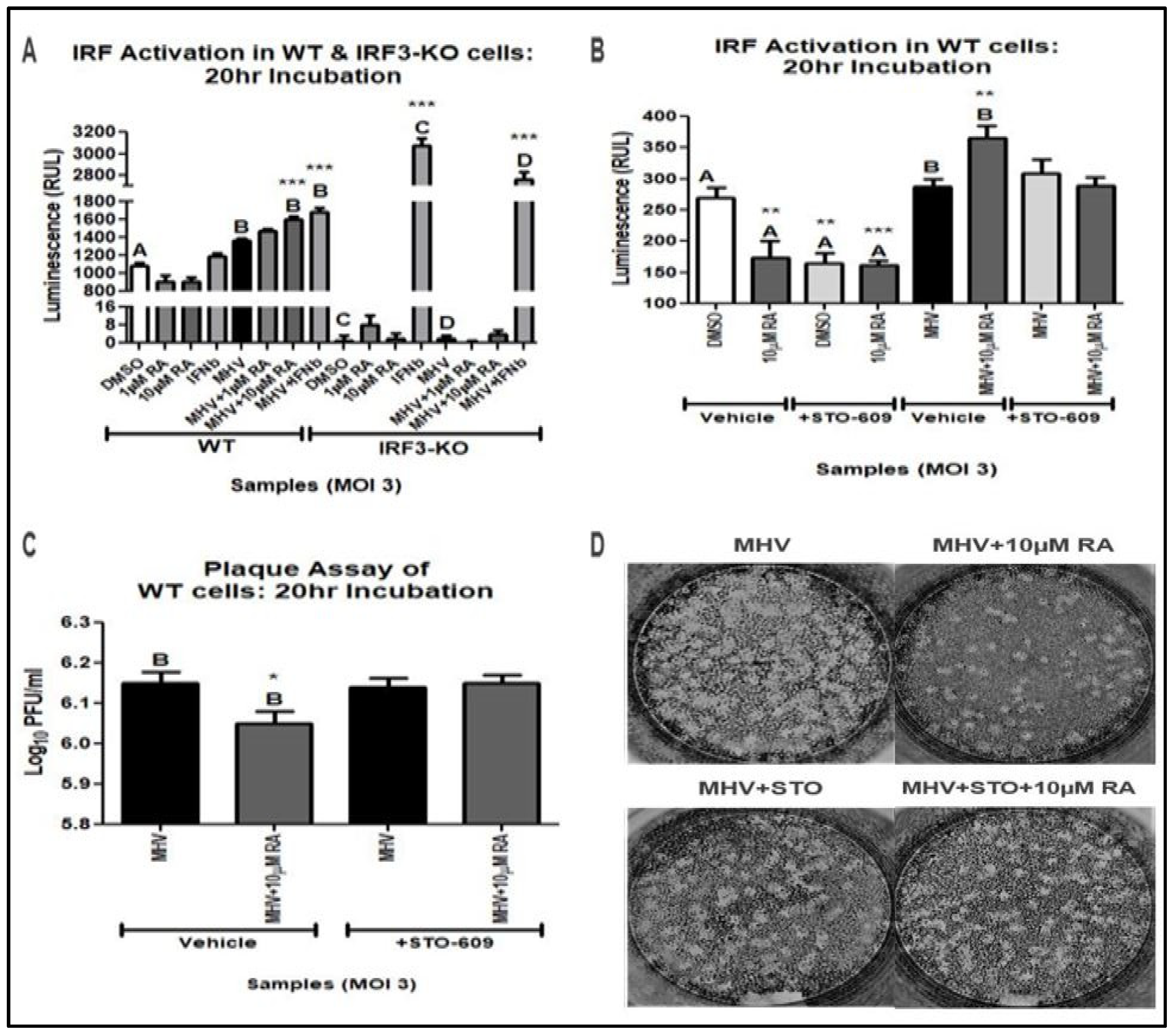
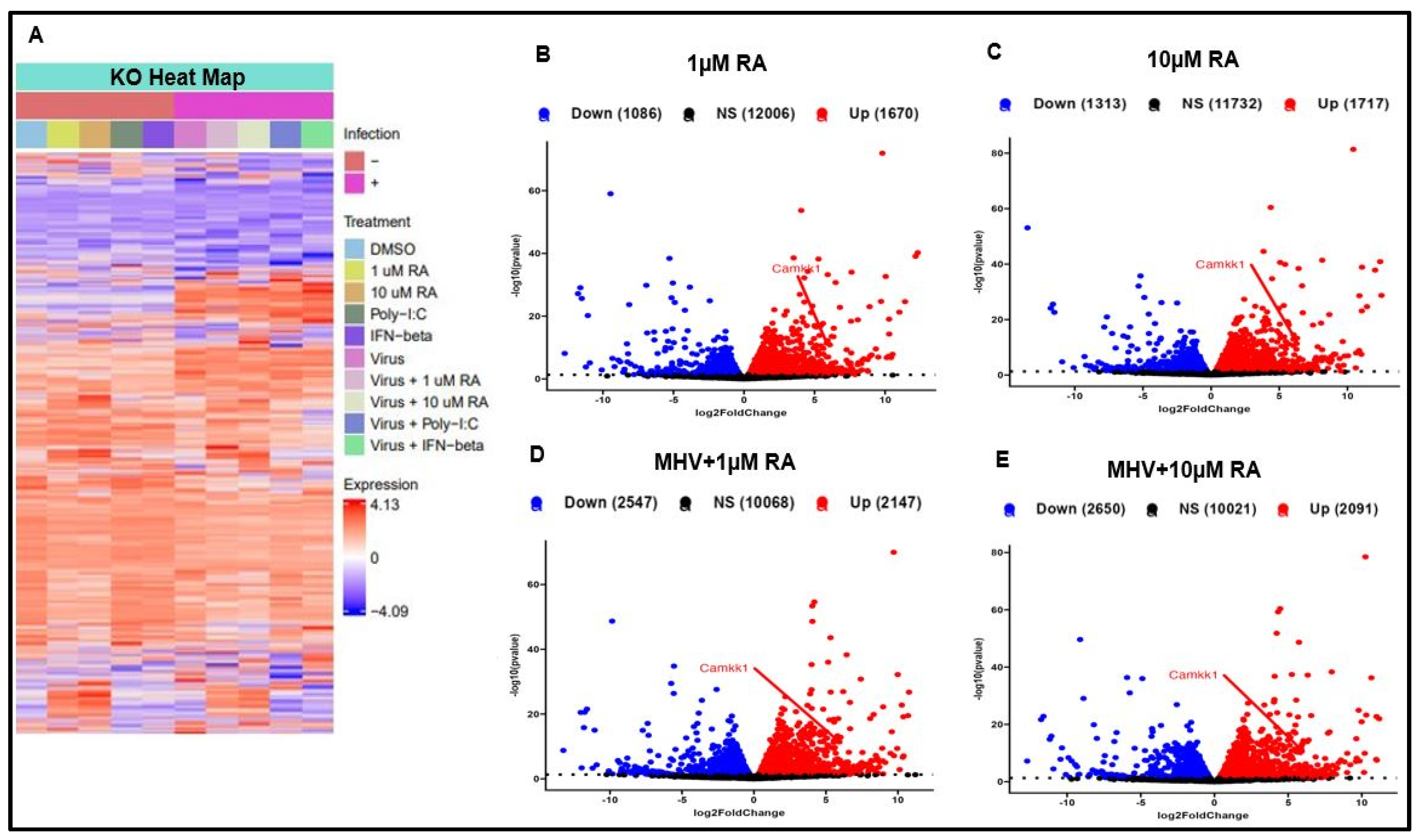
3.2. IRF3 Is Required for RA-Induced MHV Suppression
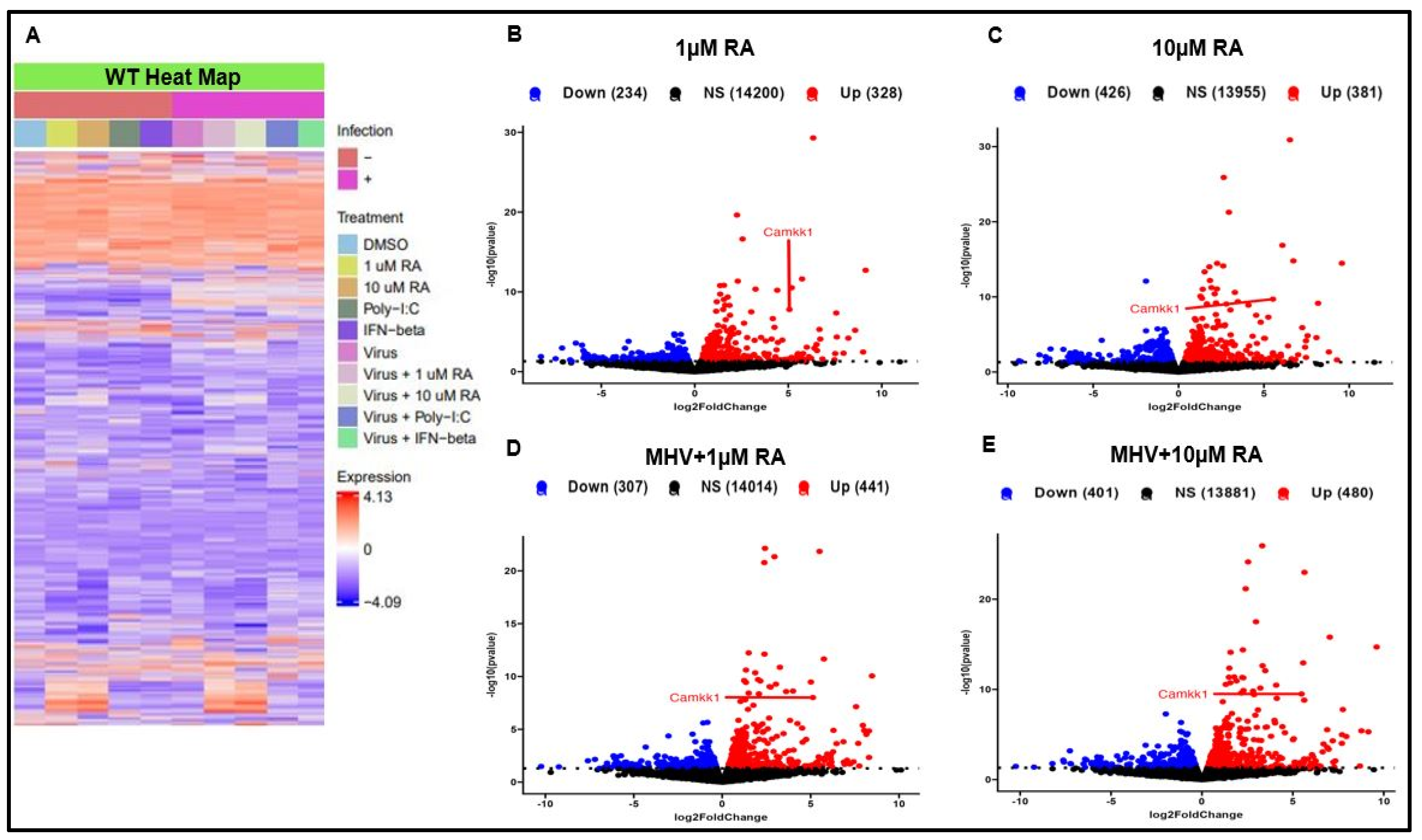
3.3. RA Promotes Activation of the Type-I IFN Response via IRF3 and CaMKK
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolf, J.M.; Wolf, L.M.; Bello, G.L.; Maccari, J.G.; Nasi, L.A. Molecular evolution of SARS-CoV-2 from December 2019 to August 2022. J. Med. Virol. 2023, 95, e28366. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Merad, M.; Blish, C.A.; Sallusto, F.; Iwasaki, A. The immunology and immunopathology of COVID-19. Science 2022, 375, 1122–1127. [Google Scholar] [CrossRef]
- Franco, J.H.; Chattopadhyay, S.; Pan, Z.K. How Different Pathologies Are Affected by IFIT Expression. Viruses 2023, 15, 342. [Google Scholar] [CrossRef]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 2021, 13, 575. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef]
- Popli, S.; Chakravarty, S.; Fan, S.; Glanz, A.; Aras, S.; Nagy, L.E.; Sen, G.C.; Chakravarti, R.; Chattopadhyay, S. IRF3 inhibits nuclear translocation of NF-kappaB to prevent viral inflammation. Proc. Natl. Acad. Sci. USA 2022, 119, e2121385119. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xiao, F.; Hu, D.; Ge, W.; Tian, M.; Wang, W.; Pan, P.; Wu, K.; Wu, J. SARS-CoV-2 Nucleocapsid Protein Interacts with RIG-I and Represses RIG-Mediated IFN-beta Production. Viruses 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Liu, H.M.; Chang, M.F.; Chang, S.C. Middle East Respiratory Syndrome Coronavirus Nucleocapsid Protein Suppresses Type I and Type III Interferon Induction by Targeting RIG-I Signaling. J. Virol. 2020, 94, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Fang, L.; Yuan, S.; Zhao, L.; Wang, X.; Long, S.; Wang, M.; Wang, D.; Foda, M.F.; Xiao, S. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-beta antagonizing mechanism: Attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 2017, 8, 49655–49670. [Google Scholar] [CrossRef]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Grana, C.; Ghosn, L.; Evrenoglou, T.; Jarde, A.; Minozzi, S.; Bergman, H.; Buckley, B.S.; Probyn, K.; Villanueva, G.; Henschke, N.; et al. Efficacy and safety of COVID-19 vaccines. Cochrane Database Syst. Rev. 2022, 12, CD015477. [Google Scholar] [CrossRef]
- Gulick, R.M.; Masure, H.; Pau, A.K.; Aberg, J.; Adimora, A.; Baker, J.; Kreuziger, L.B.; Bedimo, R.; Belperio, P.S.; Bhalla, A.; et al. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. Natl. Inst. Health 2023, 1, 469. [Google Scholar]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Njar, V.C.; Gediya, L.; Purushottamachar, P.; Chopra, P.; Vasaitis, T.S.; Khandelwal, A.; Mehta, J.; Huynh, C.; Belosay, A.; Patel, J. Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg. Med. Chem. 2006, 14, 4323–4340. [Google Scholar] [CrossRef]
- Pino-Lagos, K.; Guo, Y.; Noelle, R.J. Retinoic acid: A key player in immunity. Biofactors 2010, 36, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Soye, K.J.; Trottier, C.; Richardson, C.D.; Ward, B.J.; Miller, W.H., Jr. RIG-I is required for the inhibition of measles virus by retinoids. PLoS ONE 2011, 6, e22323. [Google Scholar] [CrossRef]
- Soye, K.J.; Trottier, C.; Di Lenardo, T.Z.; Restori, K.H.; Reichman, L.; Miller, W.H., Jr.; Ward, B.J. In vitro inhibition of mumps virus by retinoids. Virol. J. 2013, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, S.; Fukuda, R.; Ishimura, N.; Rumi, M.A.; Kazumori, H.; Uchida, Y.; Kadowaki, Y.; Ishihara, S.; Kinoshita, Y. 9-cis retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor. J. Lab. Clin. Med. 2003, 141, 58–66. [Google Scholar] [CrossRef]
- Maeda, Y.; Yamaguchi, T.; Hijikata, Y.; Morita, Y.; Tanaka, M.; Hirase, C.; Takai, S.; Tatsumi, Y.; Kanamaru, A. All-trans retinoic acid attacks reverse transcriptase resulting in inhibition of HIV-1 replication. Hematology 2007, 12, 263–266. [Google Scholar] [CrossRef]
- Morita, T.; Miyakawa, K.; Jeremiah, S.S.; Yamaoka, Y.; Sada, M.; Kuniyoshi, T.; Yang, J.; Kimura, H.; Ryo, A. All-Trans Retinoic Acid Exhibits Antiviral Effect against SARS-CoV-2 by Inhibiting 3CLpro Activity. Viruses 2021, 13, 1669. [Google Scholar] [CrossRef]
- Yuan, S.; Chu, H.; Chan, J.F.; Ye, Z.W.; Wen, L.; Yan, B.; Lai, P.M.; Tee, K.M.; Huang, J.; Chen, D.; et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 120. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Korner, R.W.; Majjouti, M.; Alcazar, M.A.A.; Mahabir, E. Of Mice and Men: The Coronavirus MHV and Mouse Models as a Translational Approach to Understand SARS-CoV-2. Viruses 2020, 12, 880. [Google Scholar] [CrossRef]
- Pereira Oliveira, G.; Kroon, E.G. Mouse hepatitis virus: A betacoronavirus model to study the virucidal activity of air disinfection equipment on surface contamination. J. Virol. Methods 2021, 297, 114274. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, A.B.; Brown, A.C.; Chitlapilly Dass, S. Murine Hepatitis Virus, a Biosafety Level 2 Model for SARS-CoV-2, Can Remain Viable on Meat and Meat Packaging Materials for at Least 48 Hours. Microbiol. Spectr. 2022, 10, e0186222. [Google Scholar] [CrossRef] [PubMed]
- Paidas, M.J.; Mohamed, A.B.; Norenberg, M.D.; Saad, A.; Barry, A.F.; Colon, C.; Kenyon, N.S.; Jayakumar, A.R. Multi-Organ Histopathological Changes in a Mouse Hepatitis Virus Model of COVID-19. Viruses 2021, 13, 1703. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Du, J.; Chen, G.; Zhao, J.; Yang, X.; Su, L.; Cheng, G.; Tang, H. Coronavirus MHV-A59 infects the lung and causes severe pneumonia in C57BL/6 mice. Virol. Sin. 2014, 29, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Axel, D.I.; Frigge, A.; Dittmann, J.; Runge, H.; Spyridopoulos, I.; Riessen, R.; Viebahn, R.; Karsch, K.R. All-trans retinoic acid regulates proliferation, migration, differentiation, and extracellular matrix turnover of human arterial smooth muscle cells. Cardiovasc. Res. 2001, 49, 851–862. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca(2+)/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Hook, S.S.; Means, A.R. Ca(2+)/CaM-dependent kinases: From activation to function. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 471–505. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Sakagami, H. Molecular Mechanisms Underlying Ca(2+)/Calmodulin-Dependent Protein Kinase Kinase Signal Transduction. Int. J. Mol. Sci. 2022, 23, 11025. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Guest, C.B.; Deszo, E.L.; Hartman, M.E.; York, J.M.; Kelley, K.W.; Freund, G.G. Ca2+/calmodulin-dependent kinase kinase alpha is expressed by monocytic cells and regulates the activation profile. PLoS ONE 2008, 3, e1606. [Google Scholar] [CrossRef] [PubMed]
- Austenaa, L.M.; Carlsen, H.; Hollung, K.; Blomhoff, H.K.; Blomhoff, R. Retinoic acid dampens LPS-induced NF-kappaB activity: Results from human monoblasts and in vivo imaging of NF-kappaB reporter mice. J. Nutr. Biochem. 2009, 20, 726–734. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Butchi, N.B.; Hinton, D.R.; Stohlman, S.A.; Kapil, P.; Fensterl, V.; Sen, G.C.; Bergmann, C.C. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol. 2014, 88, 1051–1064. [Google Scholar] [CrossRef]
- Das Sarma, J.; Burrows, A.; Rayman, P.; Hwang, M.H.; Kundu, S.; Sharma, N.; Bergmann, C.; Sen, G.C. Ifit2 deficiency restricts microglial activation and leukocyte migration following murine coronavirus (m-CoV) CNS infection. PLoS Pathog. 2020, 16, e1009034. [Google Scholar] [CrossRef]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- Prantner, D.; Perkins, D.J.; Vogel, S.N. AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling. J. Biol. Chem. 2017, 292, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Merhi, F.; Alvarez-Valadez, K.; Trepiana, J.; Lescoat, C.; Groppi, A.; Dupuy, J.W.; Soubeyran, P.; Kroemer, G.; Vacher, P.; Djavaheri-Mergny, M. Targeting CAMKK2 and SOC Channels as a Novel Therapeutic Approach for Sensitizing Acute Promyelocytic Leukemia Cells to All-Trans Retinoic Acid. Cells 2021, 10, 3364. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; van Dam, A.M.; Wang, Y.; Lucassen, P.J.; Zhou, J.N. Retinoic acid and depressive disorders: Evidence and possible neurobiological mechanisms. Neurosci. Biobehav. Rev. 2020, 112, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.D.; Shearer, K.D.; McCaffery, P.J. Retinoic acid and affective disorders: The evidence for an association. J. Clin. Psychiatry 2012, 73, 37–50. [Google Scholar] [CrossRef]
- Salaciak, K.; Koszalka, A.; Zmudzka, E.; Pytka, K. The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders. Int. J. Mol. Sci. 2021, 22, 4307. [Google Scholar] [CrossRef]
- Kaitsuka, T.; Li, S.T.; Nakamura, K.; Takao, K.; Miyakawa, T.; Matsushita, M. Forebrain-specific constitutively active CaMKKalpha transgenic mice show deficits in hippocampus-dependent long-term memory. Neurobiol. Learn. Mem. 2011, 96, 238–247. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | |
|---|---|---|
| Forward | Reverse | |
| Ifit1 | TTT CCG TAG GAA ACA TCG CGT | TGC TTG TAG CAG AGC CCT TTT |
| Ifit2 | AGT ACA ACG AGT AAG GAG TCA CT | AGG CCA GTA TGT TGC ACA TGG |
| Ifit3 | GAA GCT GAA GGG GAG CGA TT | TCC CTG TAA CGG CAC ATG AC |
| MHV-N | GGA CAG GGA GTG CCT ATT GC | TGG GGC CCT GTT CCA AGA TA |
| β-Actin | GGC TT ATT CCC CTC ATT GC | CCA GTT GGT AAC AAT GCC ATG T |
| 18s rRNA | CCG CGG TTC TAT TTT GTT GGT | CTC TAG CGG CGC AAT ACG A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, J.H.; Harris, R.A.; Ryan, W.G.; Taylor, R.T.; McCullumsmith, R.E.; Chattopadhyay, S.; Pan, Z.K. Retinoic Acid-Mediated Inhibition of Mouse Coronavirus Replication Is Dependent on IRF3 and CaMKK. Viruses 2024, 16, 140. https://doi.org/10.3390/v16010140
Franco JH, Harris RA, Ryan WG, Taylor RT, McCullumsmith RE, Chattopadhyay S, Pan ZK. Retinoic Acid-Mediated Inhibition of Mouse Coronavirus Replication Is Dependent on IRF3 and CaMKK. Viruses. 2024; 16(1):140. https://doi.org/10.3390/v16010140
Chicago/Turabian StyleFranco, Justin H., Ryan A. Harris, William G. Ryan, Roger Travis Taylor, Robert E. McCullumsmith, Saurabh Chattopadhyay, and Zhixing K. Pan. 2024. "Retinoic Acid-Mediated Inhibition of Mouse Coronavirus Replication Is Dependent on IRF3 and CaMKK" Viruses 16, no. 1: 140. https://doi.org/10.3390/v16010140
APA StyleFranco, J. H., Harris, R. A., Ryan, W. G., Taylor, R. T., McCullumsmith, R. E., Chattopadhyay, S., & Pan, Z. K. (2024). Retinoic Acid-Mediated Inhibition of Mouse Coronavirus Replication Is Dependent on IRF3 and CaMKK. Viruses, 16(1), 140. https://doi.org/10.3390/v16010140