
Identification and Genome Characterization of a Novel Virus within the Genus Totivirus from Chinese Bayberry (Myrica rubra)

Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Sample Collection and RNA-Seq Sequencing Preparation
2.2. RNA Sequencing and De Novo Transcriptome Assembly
2.3. Viral Contigs Identification, Calculations
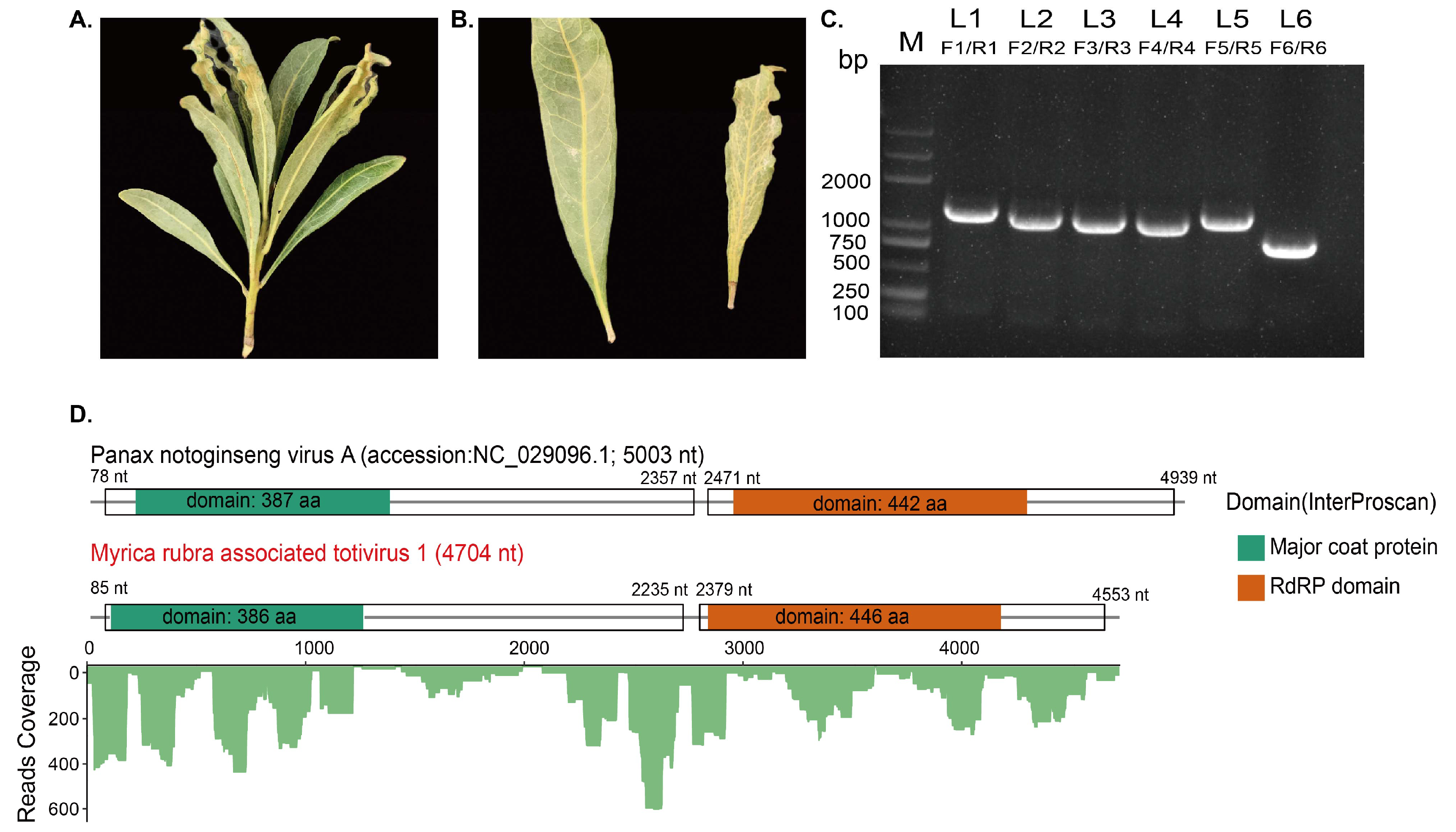
2.4. Overlapping RT-PCR and RACE
2.5. Viral Sequence Analysis
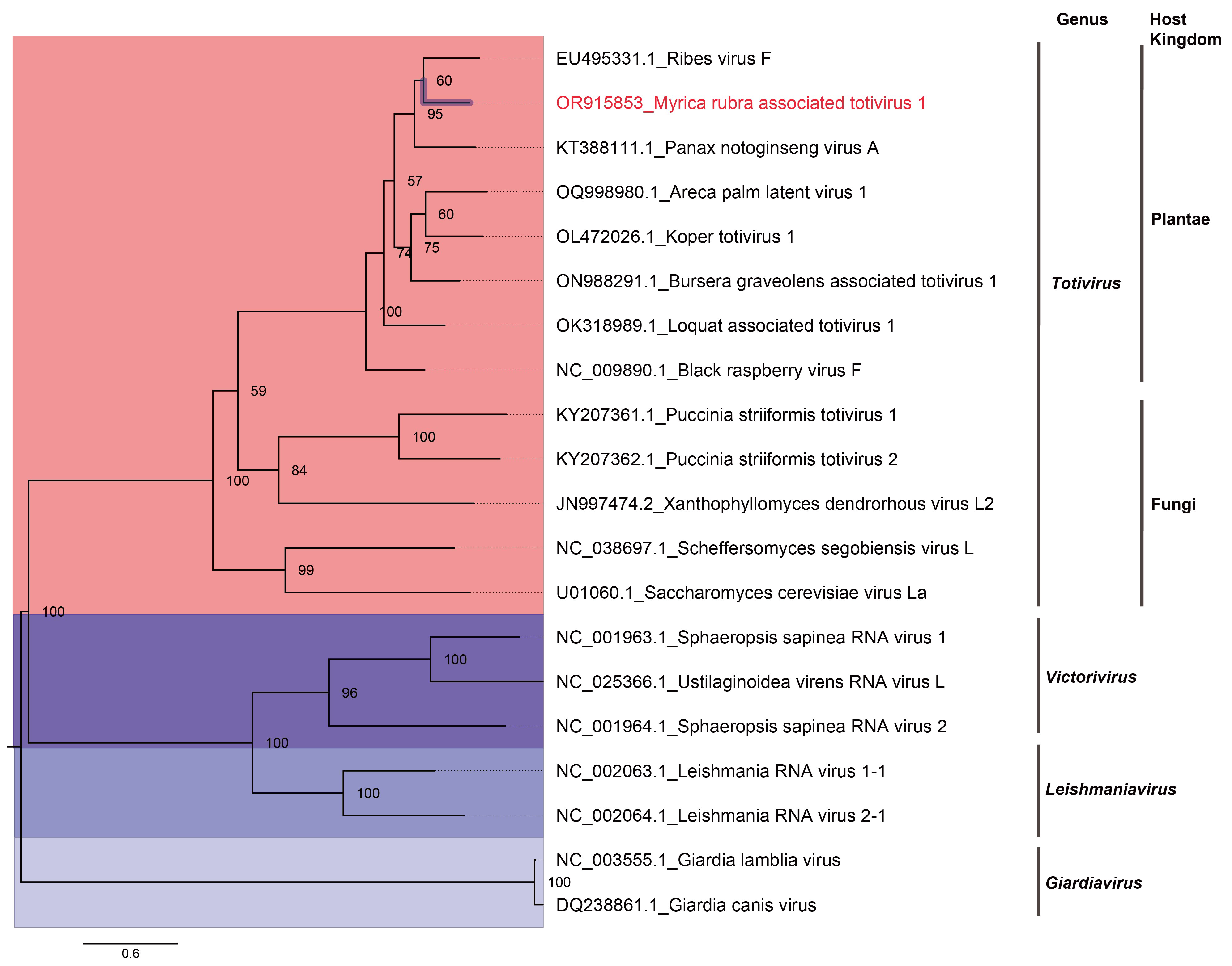
2.6. Construction of Phylogenetic Trees
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, H.; Yu, H.; Zhang, S.; Liang, S.; Zheng, X.; Zhang, S.; Yao, P.; Zheng, H.; Qi, X. Genome sequencing provides insights into the evolution and antioxidant activity of Chinese bayberry. BMC Genom. 2019, 20, 458. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Naidu, R.A. Global dimensions of plant virus diseases: Current status and future perspectives. Annu. Rev. Virol. 2019, 6, 387–409. [Google Scholar] [CrossRef]
- Gao, L.; Anane, R.F.; Chen, Z.; He, Y.; Li, S.; Zi, S.; Yang, Z.; Chu, B.; Wen, G.; Zhao, M. Complete genome sequence analysis of a novel citlodavirus isolated from the leaves of Myrica rubra in Yunnan. Arch. Virol. 2023, 168, 139. [Google Scholar] [CrossRef]
- Zhai, Y.; Attoui, H.; Mohd Jaafar, F.; Wang, H.Q.; Cao, Y.X.; Fan, S.P.; Sun, Y.X.; Liu, L.D.; Mertens, P.P.; Meng, W.S.; et al. Isolation and full-length sequence analysis of Armigeres subalbatus totivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J. Gen. Virol. 2010, 91, 2836–2845. [Google Scholar] [CrossRef]
- Guo, L.; Yang, X.; Wu, W.; Tan, G.; Fang, S.; Zhang, S.; Li, F. Identification and molecular characterization of Panax notoginseng Virus A, which may represent an undescribed novel species of the genus Totivirus, family Totiviridae. Arch. Virol. 2016, 161, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, M.E.; MacDiarmid, R.M. Molecular characterization of two totiviruses from the commensal yeast Geotrichum candidum. Viruses 2023, 15, 2150. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Creagh, J.W.; Fredericks, L.R.; Crabtree, A.M.; Patel, J.S.; Rowley, P.A. The characterization of a novel virus discovered in the yeast Pichia membranifaciens. Viruses 2022, 14, 594. [Google Scholar] [CrossRef] [PubMed]
- Isawa, H.; Kuwata, R.; Hoshino, K.; Tsuda, Y.; Sakai, K.; Watanabe, S.; Nishimura, M.; Satho, T.; Kataoka, M.; Nagata, N.; et al. Identification and molecular characterization of a new nonsegmented double-stranded RNA virus isolated from culex mosquitoes in Japan. Virus Res. 2011, 155, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Urayama, S.; Ohmatsu, T.; Sassa, Y.; Sakai, C.; Takata, M.; Hayashi, S.; Nagai, M.; Furuya, T.; Moriyama, H.; et al. Identification, Characterization and full-Length sequence analysis of a novel dsRNA virus isolated from the arboreal ant Camponotus yamaokai. J. Gen. Virol. 2015, 96, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Haugland, O.; Mikalsen, A.B.; Nilsen, P.; Lindmo, K.; Thu, B.J.; Eliassen, T.M.; Roos, N.; Rode, M.; Evensen, O. Cardiomyopathy syndrome of Atlantic salmon (Salmo salar L.) is caused by a double-stranded RNA Virus of the Totiviridae family. J. Virol. 2011, 85, 5275–5286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xu, L.; Bowers, H.; Schott, E.J. Characterization of two novel toti-like viruses co-infecting the Atlantic blue crab, callinectes sapidus, in its northern range of the United States. Front. Microbiol. 2022, 13, 855750. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Quinto, R.A.; Espinoza-Lozano, R.F.; Mora-Pinargote, C.A.; Quito-Avila, D.F. Complete genome sequence of a variant of maize-associated totivirus from Ecuador. Arch. Virol. 2017, 162, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Togoobat, B.; Wu, N.; Wang, X.; Cao, M.; Xu, Z. Viromic approach reveals differences in the composition, diversity and relative abundance of pumpkin viruses across main growing regions of China. Virology 2023, 585, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pu, Z.; Ni, H.; Wang, Y.; Yan, B. Multiple mycoviruses identified in Pestalotiopsis spp. from Chinese bayberry. Virol. J. 2021, 18, 43. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus | Genome (nt) | CP (aa) | RdRP (aa) | 5′UTR (nt) | 3′UTR (nt) | IR 1 (nt) | GC (%) |
|---|---|---|---|---|---|---|---|
| MRaTV1 | 4704 | 716 | 724 | 84 | 151 | 126 | 51.15 |
| 2 PnVA | 5003 | 760 | 823 | 77 | 64 | 114 | 43.11 |
| 3 BgTV1 | 4794 | 687 | 767 | 67 | 57 | 309 | 43.14 |
| 4 BrVF | 5077 | 770 | 815 | 198 | 78 | 147 | 45.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Gao, Y.; Teng, K.; Ge, H.; Zhang, X.; Wu, M.; Li, R.; Wu, Z.; Zheng, L. Identification and Genome Characterization of a Novel Virus within the Genus Totivirus from Chinese Bayberry (Myrica rubra). Viruses 2024, 16, 283. https://doi.org/10.3390/v16020283
Xu Z, Gao Y, Teng K, Ge H, Zhang X, Wu M, Li R, Wu Z, Zheng L. Identification and Genome Characterization of a Novel Virus within the Genus Totivirus from Chinese Bayberry (Myrica rubra). Viruses. 2024; 16(2):283. https://doi.org/10.3390/v16020283
Chicago/Turabian StyleXu, Zhongtian, Yi’nan Gao, Kun Teng, Huoyang Ge, Xiaoqi Zhang, Mengjing Wu, Ruhui Li, Zujian Wu, and Luping Zheng. 2024. "Identification and Genome Characterization of a Novel Virus within the Genus Totivirus from Chinese Bayberry (Myrica rubra)" Viruses 16, no. 2: 283. https://doi.org/10.3390/v16020283
APA StyleXu, Z., Gao, Y., Teng, K., Ge, H., Zhang, X., Wu, M., Li, R., Wu, Z., & Zheng, L. (2024). Identification and Genome Characterization of a Novel Virus within the Genus Totivirus from Chinese Bayberry (Myrica rubra). Viruses, 16(2), 283. https://doi.org/10.3390/v16020283