
Epidemiological and Genetic Characterization of Coxsackievirus A6-Associated Hand, Foot, and Mouth Disease in Gwangju, South Korea, in 2022

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection
2.2. Detection and Identification of CV-A6
2.3. Complete Genome Sequencing
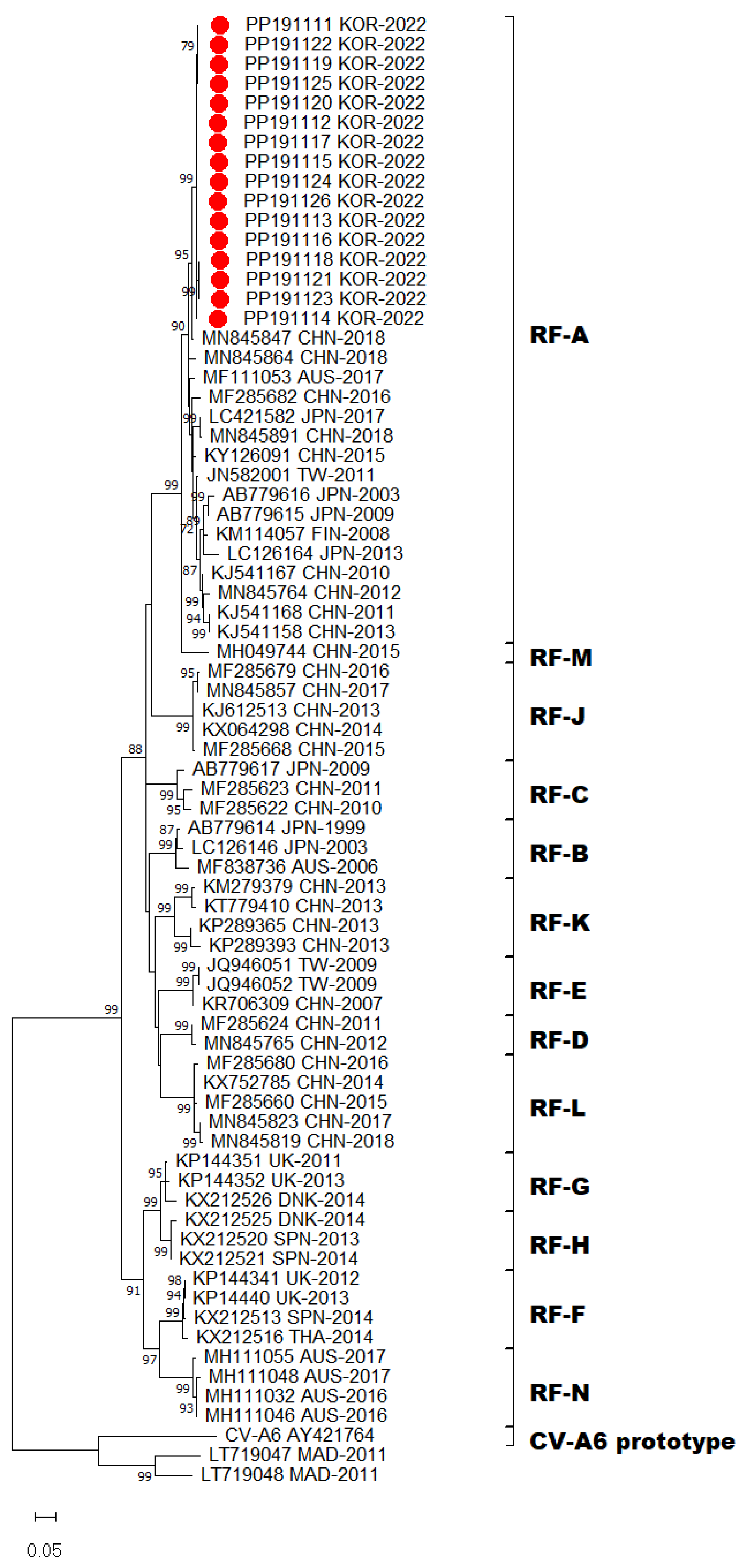
2.4. Phylogenetic Analyses
2.5. Mutation Analyses
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oberste, M.S.; Peñaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species Human enterovirus A. J. Gen. Virol. 2004, 85, 1597–1607. [Google Scholar] [PubMed]
- Zhang, Y.; Tan, X.-J.; Wang, H.-Y.; Yan, D.-M.; Zhu, S.-L.; Wang, D.-Y.; Ji, F.; Wang, X.-J.; Gao, Y.-J.; Chen, L. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 2009, 44, 262–267. [Google Scholar]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y. Hand, foot, and mouth disease in China, 2008–2012: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar]
- Zhang, Y.; Wang, D.; Yan, D.; Zhu, S.; Liu, J.; Wang, H.; Zhao, S.; Yu, D.; Nan, L.; An, J. Molecular evidence of persistent epidemic and evolution of subgenotype B1 coxsackievirus A16-associated hand, foot, and mouth disease in China. J. Clin. Microbiol. 2010, 48, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Österback, R.; Vuorinen, T.; Linna, M.; Susi, P.; Hyypiä, T.; Waris, M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg. Infect. Dis. 2009, 15, 1485. [Google Scholar] [PubMed]
- Montes, M.; Artieda, J.; Pineiro, L.D.; Gastesi, M.; Diez-Nieves, I.; Cilla, G. Hand, foot, and mouth disease outbreak and coxsackievirus A6, northern Spain, 2011. Emerg. Infect. Dis. 2013, 19, 676. [Google Scholar] [PubMed]
- Sinclair, C.; Gaunt, E.; Simmonds, P.; Broomfield, D.; Nwafor, N.; Wellington, L.; Templeton, K.; Willocks, L.; Schofield, O.; Harvala, H. Atypical hand, foot, and mouth disease associated with coxsackievirus A6 infection, Edinburgh, United Kingdom, January to February 2014. Eurosurveillance 2014, 19, 20745. [Google Scholar] [PubMed]
- Tomba Ngangas, S.; Bisseux, M.; Jugie, G.; Lambert, C.; Cohen, R.; Werner, A.; Archimbaud, C.; Henquell, C.; Mirand, A.; Bailly, J.-L. Coxsackievirus A6 recombinant subclades D3/A and D3/H were predominant in hand-foot-and-mouth disease outbreaks in the paediatric population, France, 2010–2018. Viruses 2022, 14, 1078. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Notes from the field: Severe hand, foot, and mouth disease associated with coxsackievirus A6-Alabama, Connecticut, California, and Nevada, November 2011–February 2012. MMWR. Morb. Mortal. Wkly. Rep. 2012, 61, 213–214. [Google Scholar]
- Wu, Y.; Yeo, A.; Phoon, M.; Tan, E.; Poh, C.; Quak, S.; Chow, V.T. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: The role of enterovirus 71 and coxsackievirus A strains. Int. J. Infect. Dis. 2010, 14, e1076–e1081. [Google Scholar] [CrossRef]
- Lau, S.K.; Zhao, P.S.; Sridhar, S.; Yip, C.C.; Aw-Yong, K.L.; Chow, E.Y.; Cheung, K.C.; Hui, R.W.; Leung, R.Y.; Lai, Y.S. Molecular epidemiology of coxsackievirus A6 circulating in Hong Kong reveals common neurological manifestations and emergence of novel recombinant groups. J. Clin. Virol. 2018, 108, 43–49. [Google Scholar]
- Fujimoto, T.; Iizuka, S.; Enomoto, M.; Abe, K.; Yamashita, K.; Hanaoka, N.; Okabe, N.; Yoshida, H.; Yasui, Y.; Kobayashi, M. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg. Infect. Dis. 2012, 18, 337. [Google Scholar]
- Puenpa, J.; Chieochansin, T.; Linsuwanon, P.; Korkong, S.; Thongkomplew, S.; Vichaiwattana, P.; Theamboonlers, A.; Poovorawan, Y. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg. Infect. Dis. 2013, 19, 641. [Google Scholar] [PubMed]
- Puenpa, J.; Saengdao, N.; Khanarat, N.; Korkong, S.; Chansaenroj, J.; Yorsaeng, R.; Wanlapakorn, N.; Poovorawan, Y. Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022. Viruses 2022, 15, 73. [Google Scholar] [PubMed]
- Han, J.-F.; Xu, S.; Zhang, Y.; Zhu, S.-Y.; Wu, D.-L.; Yang, X.-D.; Liu, H.; Sun, B.-X.; Wu, X.-Y.; Qin, C.-F. Hand, foot, and mouth disease outbreak caused by coxsackievirus A6, China, 2013. J. Infect. 2014, 69, 303–305. [Google Scholar] [PubMed]
- Zeng, H.; Lu, J.; Zheng, H.; Yi, L.; Guo, X.; Liu, L.; Rutherford, S.; Sun, L.; Tan, X.; Li, H. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guangdong, China. Sci. Rep. 2015, 5, 10550. [Google Scholar]
- Zhang, M.; Chen, X.; Wang, W.; Li, Q.; Xie, Z. Genetic characteristics of Coxsackievirus A6 from children with hand, foot and mouth disease in Beijing, China, 2017–2019. Infect. Genet. Evol. 2022, 106, 105378. [Google Scholar] [PubMed]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci. Rep. 2017, 7, 5491. [Google Scholar] [CrossRef] [PubMed]
- Österback, R.; Koskinen, S.; Merilahti, P.; Pursiheimo, J.-P.; Blomqvist, S.; Roivainen, M.; Laiho, A.; Susi, P.; Waris, M. Genome sequence of coxsackievirus A6, isolated during a hand-foot-and-mouth disease outbreak in Finland in 2008. Genome Announc. 2014, 2, 10-1128. [Google Scholar] [CrossRef]
- Gaunt, E.; Harvala, H.; Österback, R.; Sreenu, V.B.; Thomson, E.; Waris, M.; Simmonds, P. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: A potential role of recombination in emergence and pathogenicity. J. Gen. Virol. 2015, 96, 1067. [Google Scholar]
- Puenpa, J.; Vongpunsawad, S.; Österback, R.; Waris, M.; Eriksson, E.; Albert, J.; Midgley, S.; Fischer, T.K.; Eis-Hübinger, A.M.; Cabrerizo, M. Molecular epidemiology and the evolution of human coxsackievirus A6. J. Gen. Virol. 2016, 97, 3225–3231. [Google Scholar] [CrossRef]
- Carmona, R.C.; Machado, B.C.; Reis, F.C.; Jorge, A.M.; Cilli, A.; Dias, A.M.; Morais, D.R.; Leme, L.; Ana, L.; Silva, M.R. Hand, foot, and mouth disease outbreak by Coxsackievirus A6 during COVID-19 pandemic in 2021, São Paulo, Brazil. J. Clin. Virol. 2022, 154, 105245. [Google Scholar] [CrossRef]
- Ji, T.; Han, T.; Tan, X.; Zhu, S.; Yan, D.; Yang, Q.; Song, Y.; Cui, A.; Zhang, Y.; Mao, N. Surveillance, epidemiology, and pathogen spectrum of hand, foot, and mouth disease in mainland of China from 2008 to 2017. Biosaf. Health 2019, 1, 32–40. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, J.-e.; Kim, K.G.; Park, D.W.; Cho, S.J.; Kim, T.S.; Kee, H.-y.; Kim, S.-H.; Park, H.j.; Seo, M.H. Long-term sentinel surveillance of enteroviruses in Gwangju, South Korea, 2011–2020. Sci. Rep. 2023, 13, 2798. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. Universal PCR Primers Are Critical for Direct Sequencing-Based Enterovirus Genotyping. J. Clin. Microbiol. 2017, 55, 339–340. [Google Scholar] [CrossRef]
- Chen, M.; Zuo, X.; Tan, Y.; Ju, Y.; Bi, F.; Wang, H.; Chen, M. Six amino acids of VP1 switch along with pandemic of CV-A6-associated HFMD in Guangxi, southern China, 2010–2017. J. Infect. 2019, 78, 323–337. [Google Scholar] [PubMed]
- Mirand, A.; Cohen, R.; Bisseux, M.; Tomba, S.; Sellem, F.C.; Gelbert, N.; Béchet, S.; Frandji, B.; Archimbaud, C.; Brebion, A. A large-scale outbreak of hand, foot and mouth disease, France, as at 28 September 2021. Eurosurveillance 2021, 26, 2100978. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Total (n = 277) | % | ||
|---|---|---|---|
| Gender | Male | 148 | 53.4 |
| Female | 129 | 46.6 | |
| Age group (years) | <1 | 27 | 9.7 |
| 1 to 4 | 224 | 80.9 | |
| 5 to 9 | 21 | 7.6 | |
| >9 | 5 | 1.8 | |
| Symptoms | Fever | 259 | 93.5 |
| Respiratory | 15 | 5.4 | |
| Gastrointestinal | 4 | 1.4 | |
| Unknown | 10 | 4.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-E.; Kim, M.-J.; Lim, M.-H.; Han, S.-J.; Kim, J.-Y.; Kim, S.-H.; Ha, Y.-D.; Gang, G.-L.; Chung, Y.-S.; Seo, J.-M. Epidemiological and Genetic Characterization of Coxsackievirus A6-Associated Hand, Foot, and Mouth Disease in Gwangju, South Korea, in 2022. Viruses 2024, 16, 476. https://doi.org/10.3390/v16030476
Lee J-E, Kim M-J, Lim M-H, Han S-J, Kim J-Y, Kim S-H, Ha Y-D, Gang G-L, Chung Y-S, Seo J-M. Epidemiological and Genetic Characterization of Coxsackievirus A6-Associated Hand, Foot, and Mouth Disease in Gwangju, South Korea, in 2022. Viruses. 2024; 16(3):476. https://doi.org/10.3390/v16030476
Chicago/Turabian StyleLee, Ji-Eun, Min-Ji Kim, Mi-Hyeon Lim, Sue-Ji Han, Jin-Yeong Kim, Soo-Hoo Kim, Yi-Duen Ha, Gyung-Li Gang, Yoon-Seok Chung, and Jung-Mi Seo. 2024. "Epidemiological and Genetic Characterization of Coxsackievirus A6-Associated Hand, Foot, and Mouth Disease in Gwangju, South Korea, in 2022" Viruses 16, no. 3: 476. https://doi.org/10.3390/v16030476
APA StyleLee, J.-E., Kim, M.-J., Lim, M.-H., Han, S.-J., Kim, J.-Y., Kim, S.-H., Ha, Y.-D., Gang, G.-L., Chung, Y.-S., & Seo, J.-M. (2024). Epidemiological and Genetic Characterization of Coxsackievirus A6-Associated Hand, Foot, and Mouth Disease in Gwangju, South Korea, in 2022. Viruses, 16(3), 476. https://doi.org/10.3390/v16030476