Friend Spleen Focus-Forming Virus Activates the Tyrosine Kinase sf-Stk and the Transcription Factor PU.1 to Cause a Multi-Stage Erythroleukemia in Mice

Abstract
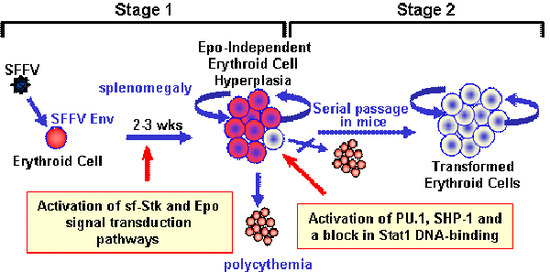
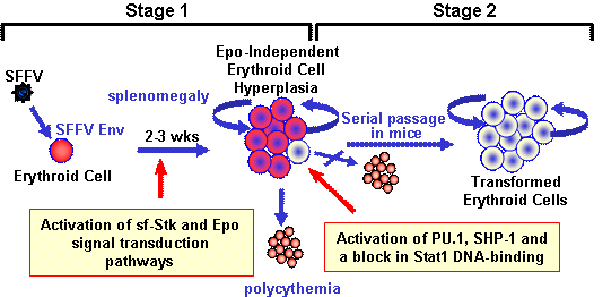
:
1. Introduction
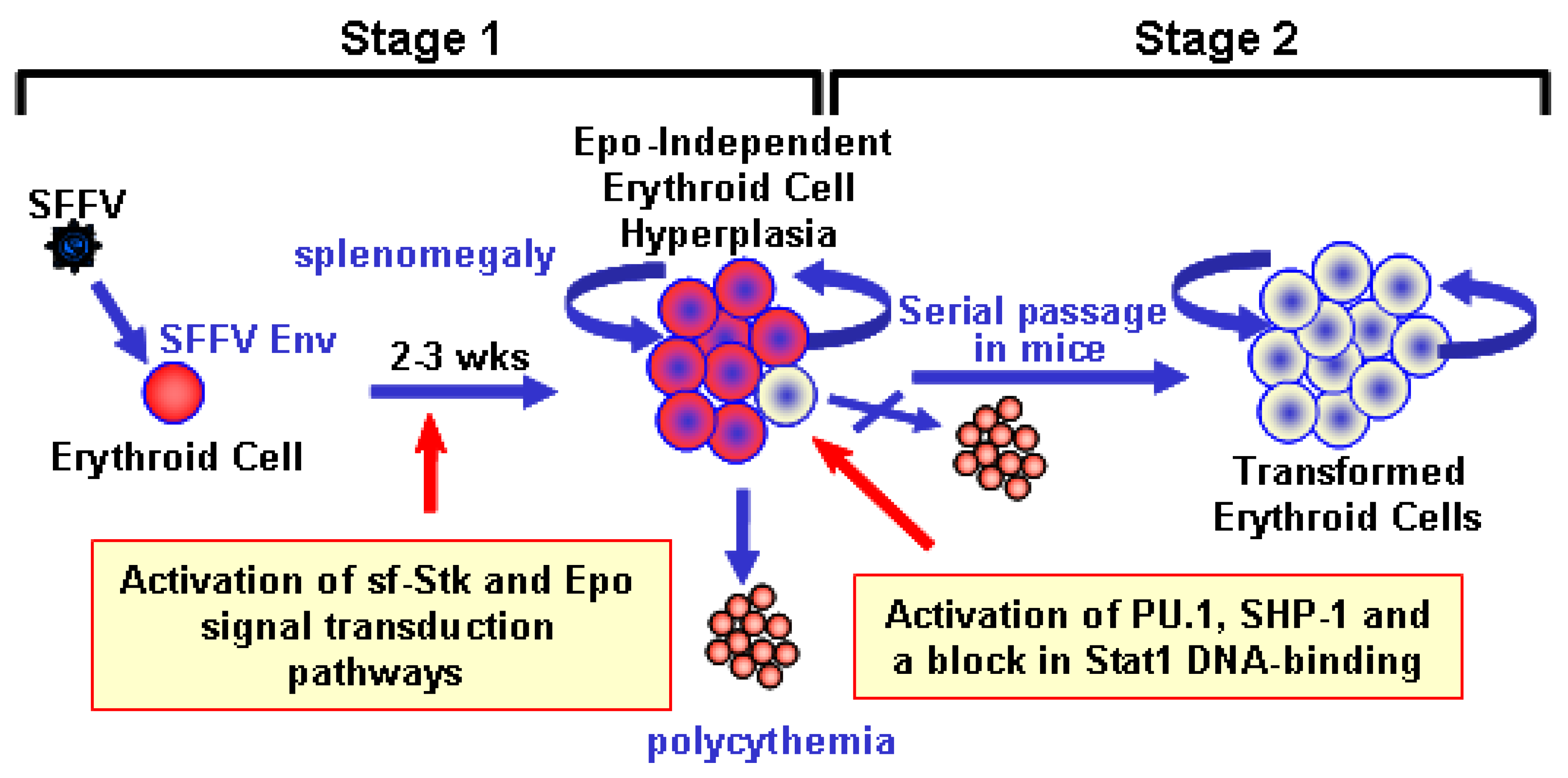
2. Biological Effects of SFFV
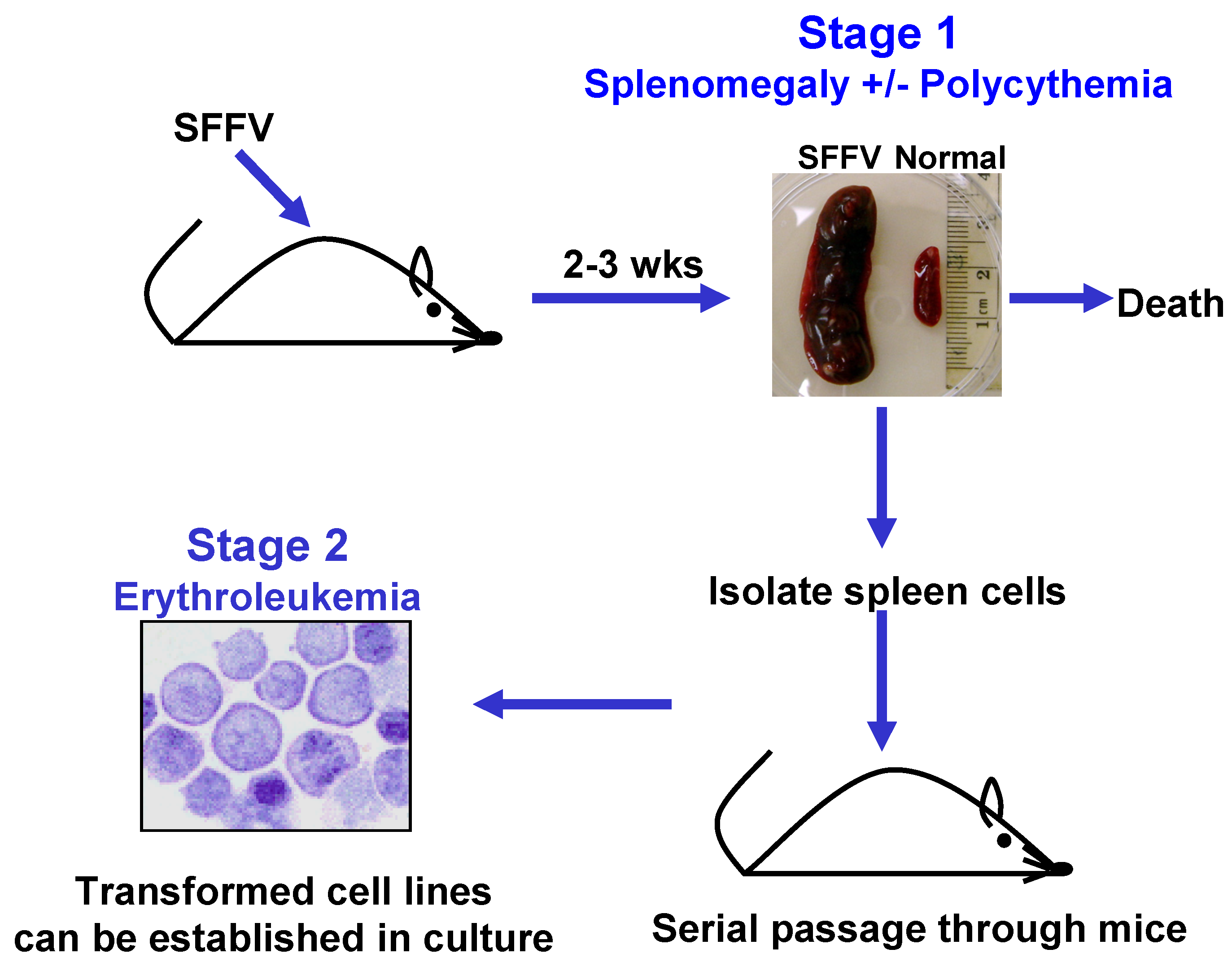
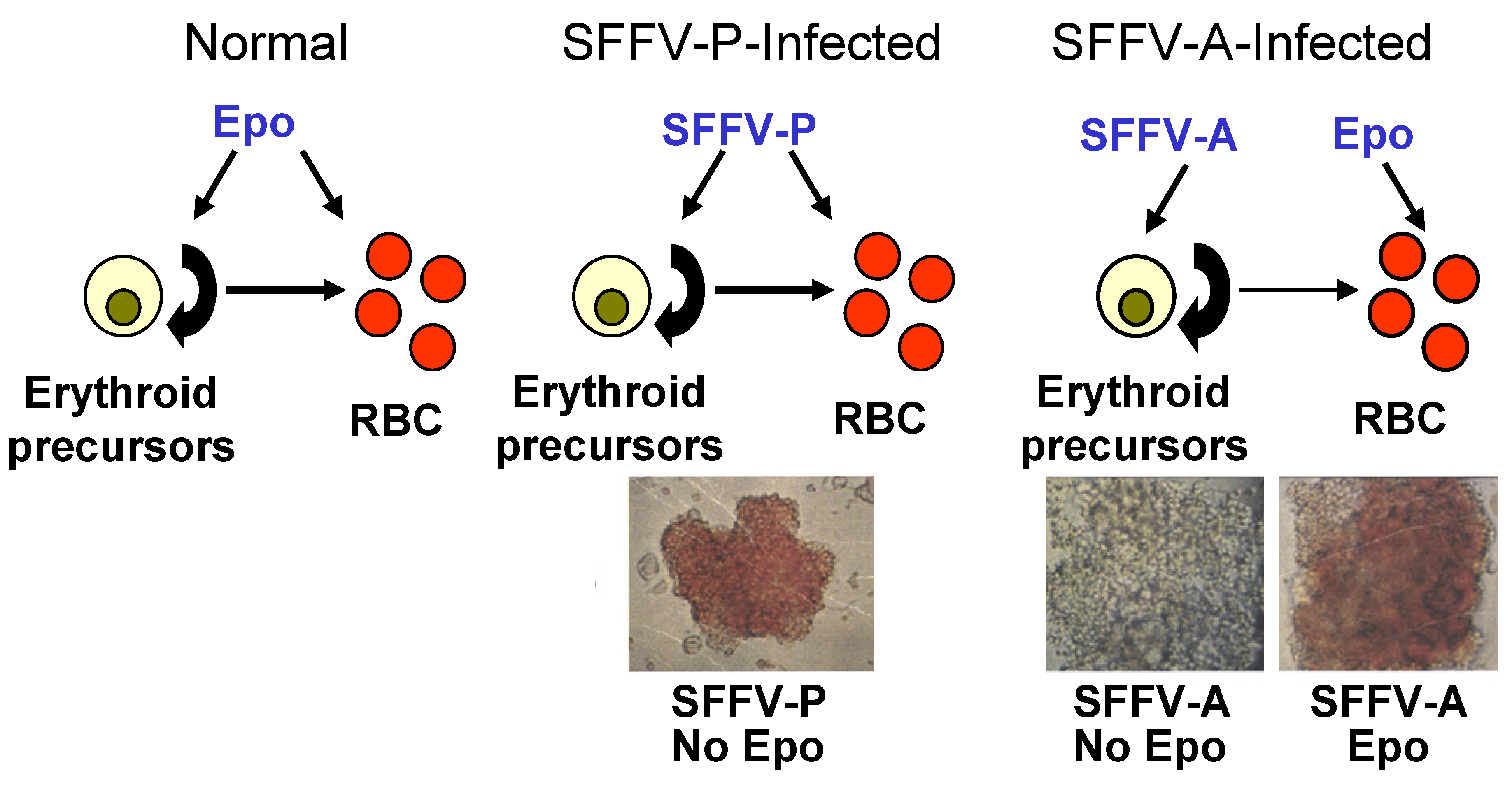
2.1. Induction of Erythroid Hyperplasia by SFFV
2.2. Transformation of Erythroid Cells by SFFV
3. Molecular Basis for the Epo-independent Erythroid Hyperplasia Induced by SFFV
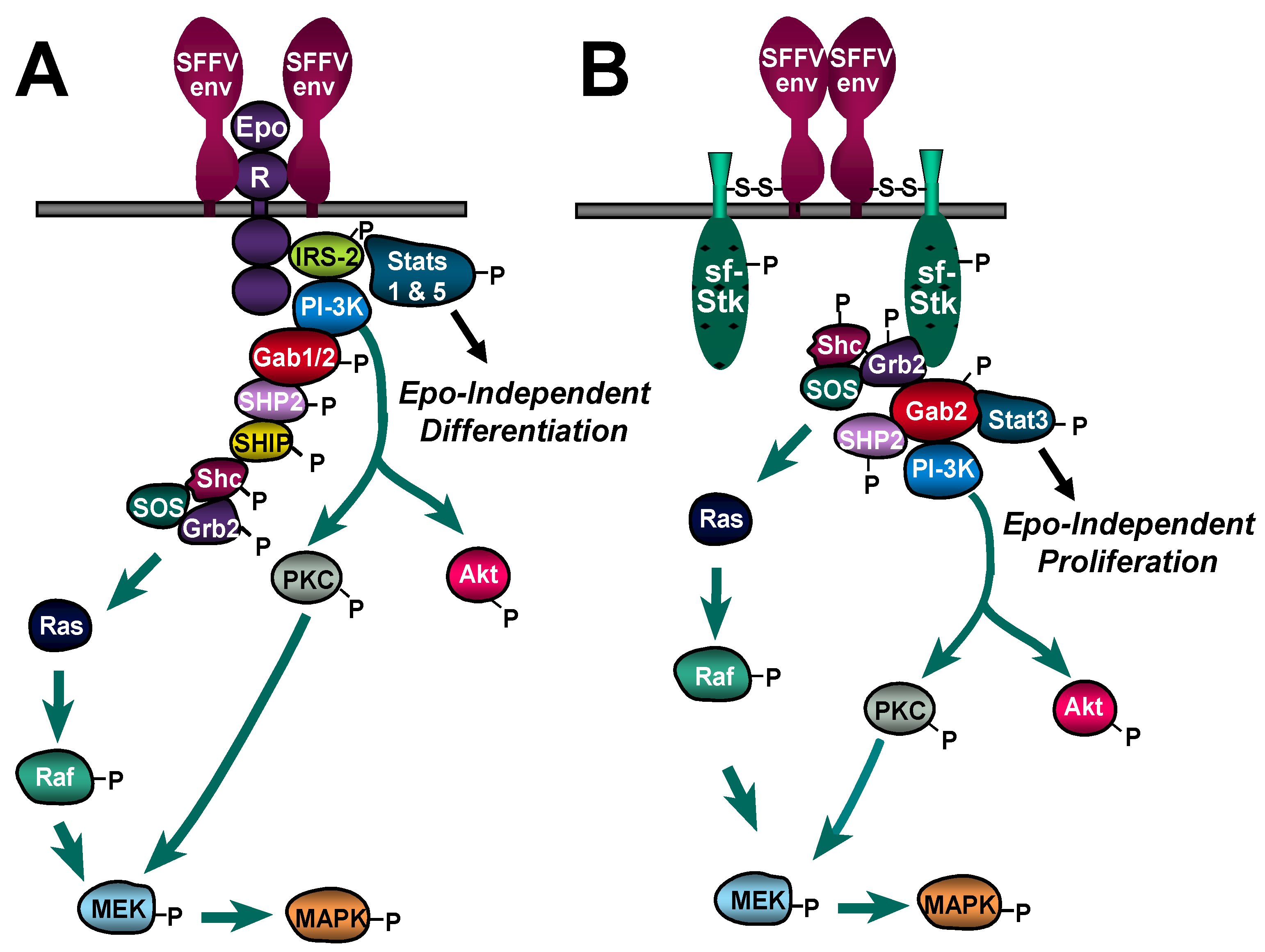
3.1. Constitutive Activation of Signal Transduction Pathways by SFFV

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Components of Epo/EpoR Pathway | SFFV-P | SFFV-A | Reference |
|---|---|---|---|
| Shc | + | + | [47] |
| Grb2 | + | + | [47] |
| Ras | + | + | [47] |
| Raf-1 | + | + | [46] |
| MEK | + | + | [46] |
| ERK 1/2 | + | + | [46] |
| JNK | + | + | [48] |
| Stat1 | + | − | [50,51,52] |
| Stat3 | + | + | [50,51,52] |
| Stat5 | + | − | [50,51,52] |
| PI 3-kinase | + | + | [43] |
| IRS-2 | + | + | [43] |
| Gab1 | + | + | [43] |
| Gab2 | + | + | [43] |
| PKC | + | + | [47] |
| Akt | + | + | [43] |
| SHIP | + | + | [43] |
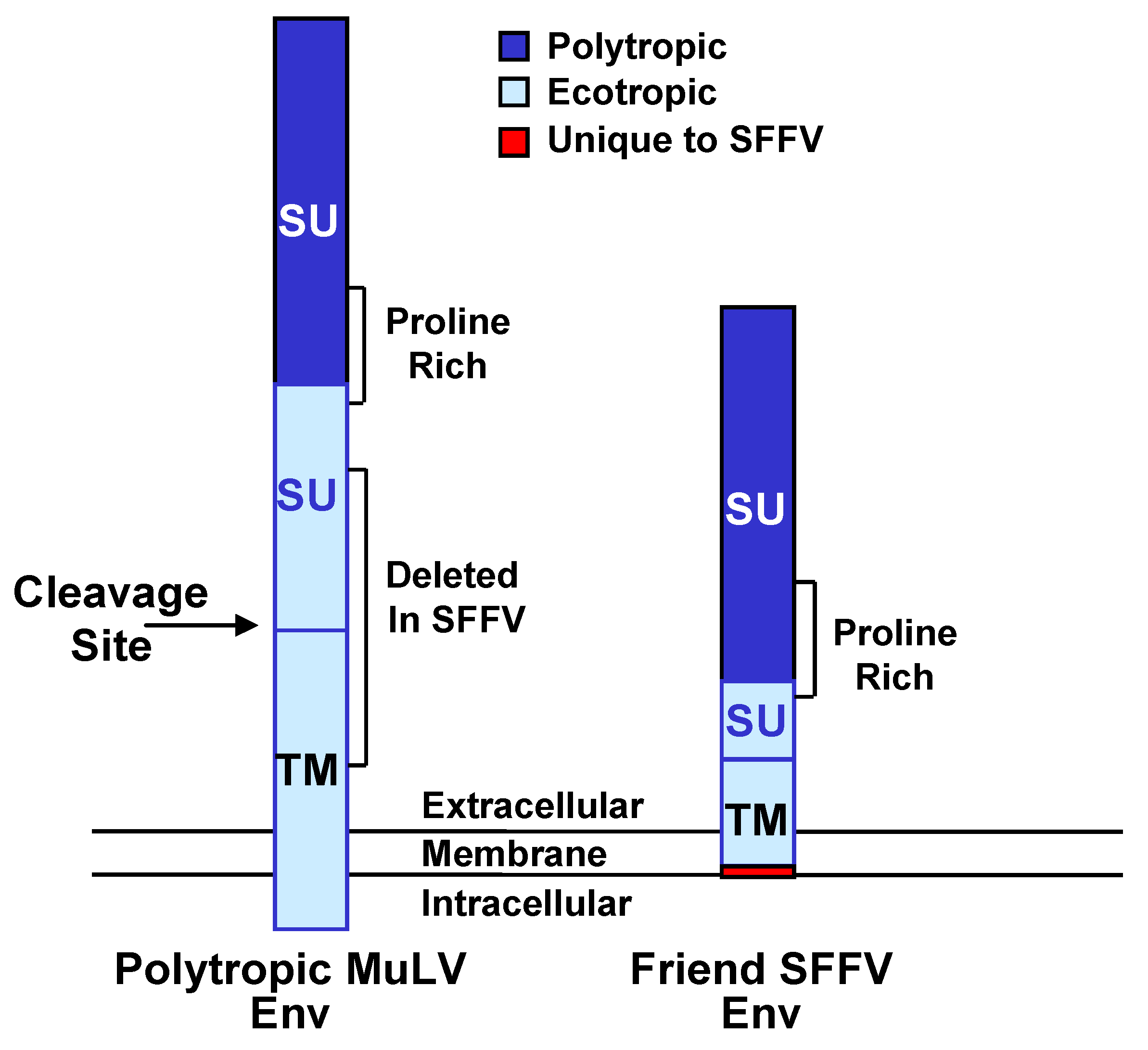
3.2. Interaction of SFFV Env with the EpoR
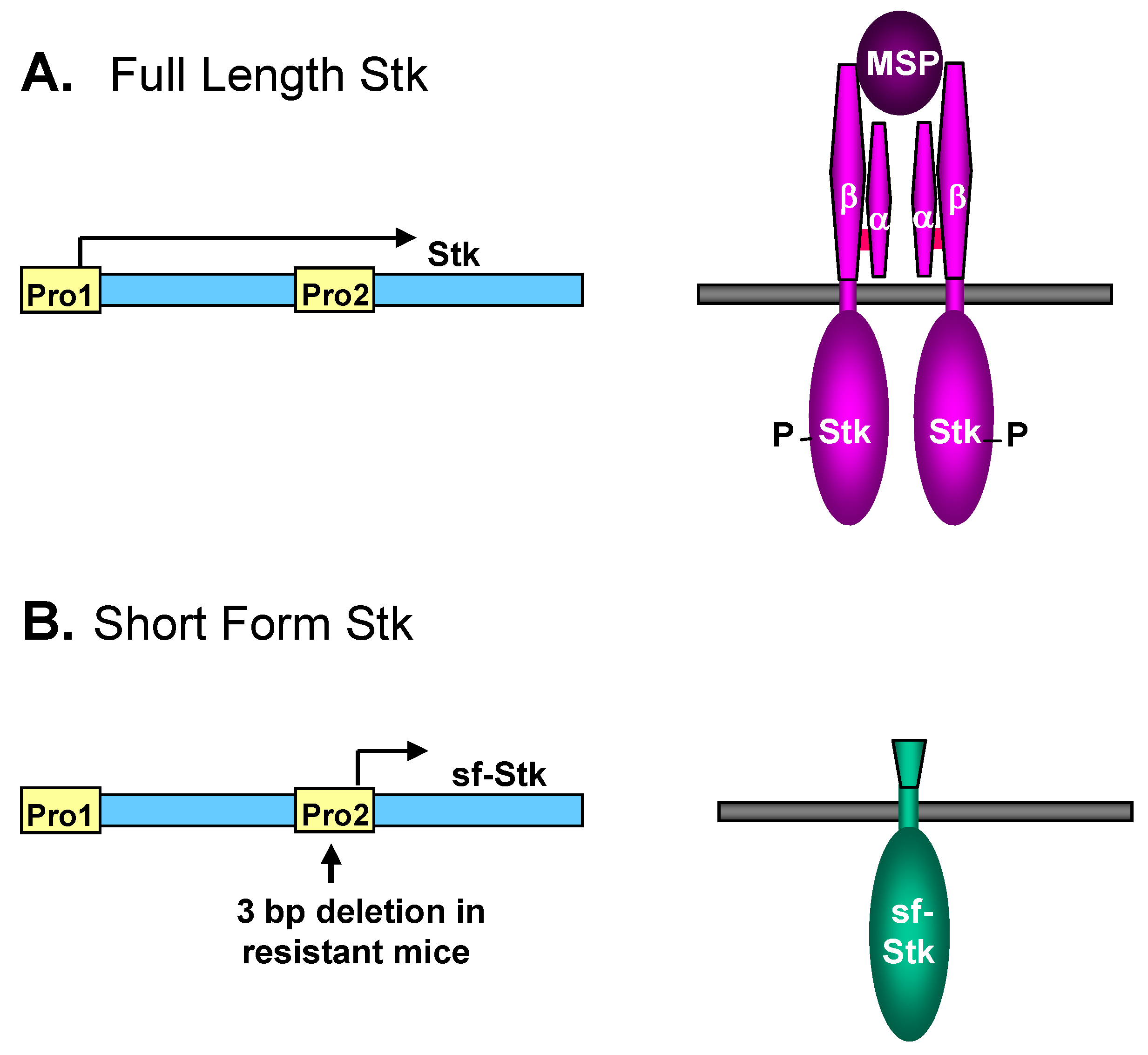
3.3. Interaction of SFFV Env with the Receptor Tyrosine Kinase sf-Stk
3.4. Different Roles of EpoR Signaling and sf-Stk in SFFV-induced Disease
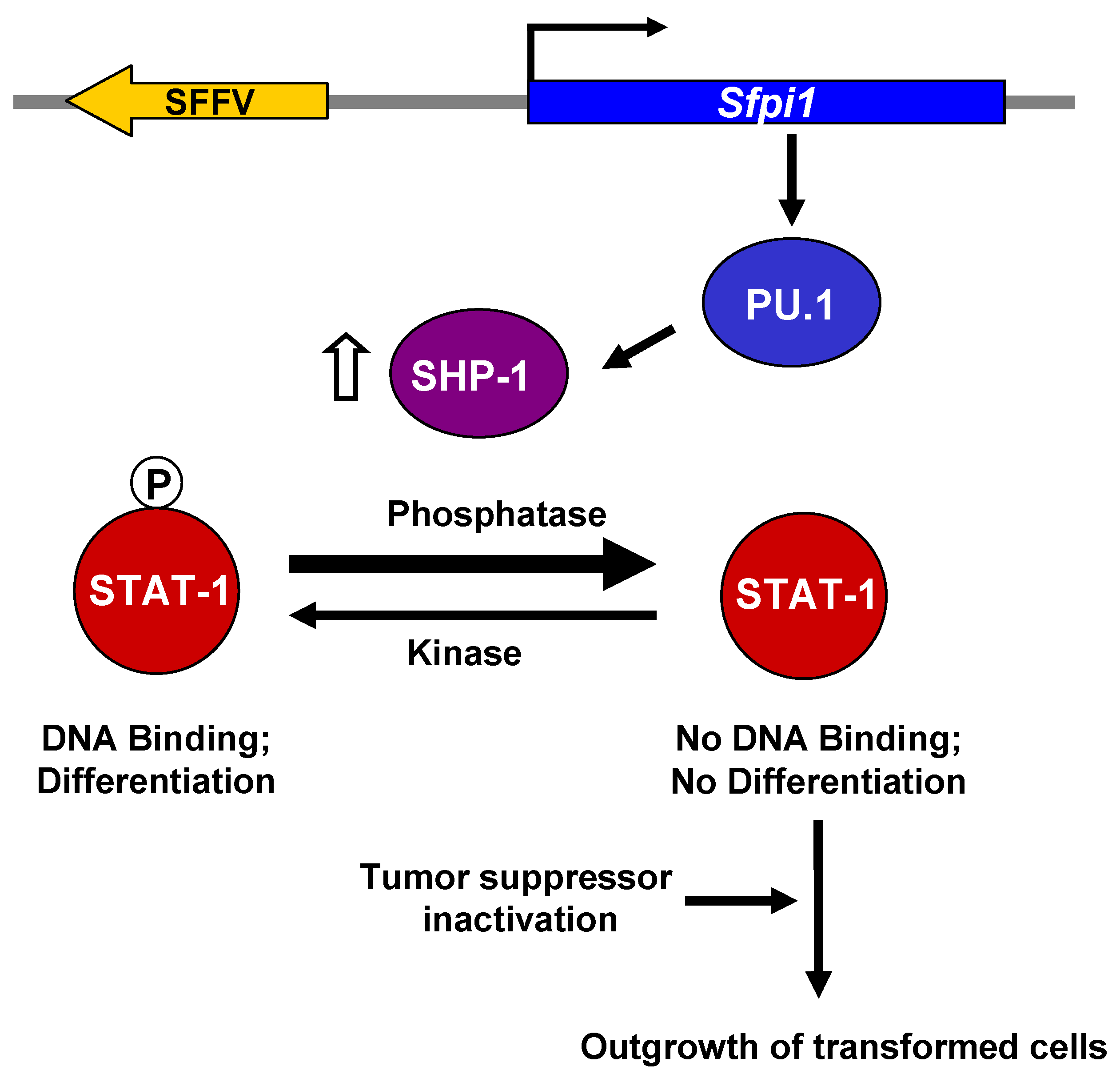
4. Molecular Basis for the Transformation of Erythroid Cells by SFFV
5. Conclusions
References and Notes
- Friend, C. Cell-Free Transmission in Adult Swiss Mice of a Disease Having the Character of a Leukemia. J. Exp. Med. 1957, 105, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, S.; Suda, Y.; Furuta, Y.; Yagi, T.; Takeda, N.; Watanabe, N.; Nagayoshi, M.; Ikawa, Y. Env-derived gp55 gene of Friend spleen focus-forming virus specifically induces neoplastic proliferation of erythroid progenitor cells. EMBO J. 1990, 9, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Ruscetti, S. The spleen focus-forming virus (SFFV) envelope gene, when introduced into mice in the absence of other SFFV genes, induces acute erythroleukemia. J. Virol. 1988, 62, 2158–2163. [Google Scholar] [CrossRef] [PubMed]
- Amanuma, H.; Katori, A.; Obata, M.; Sagata, N.; Ikawa, Y. Complete nucleotide sequence of the gene for the specific glycoprotein (gp55) of Friend spleen focus-forming virus. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 3913–3917. [Google Scholar] [CrossRef]
- Clark, S.P.; Mak, T.W. Complete nucleotide sequence of an infectious clone of Friend spleen focus-forming provirus: gp55 is an envelope fusion glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 5037–5041. [Google Scholar] [CrossRef]
- Wolff, L.; Scolnick, E.; Ruscetti, S. Envelope gene of the Friend spleen focus-forming virus: Deletion and insertions in 3’ gp70/p15E-encoding region have resulted in unique features in the primary structure of its protein product. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 4718–4722. [Google Scholar] [CrossRef]
- Amanuma, H.; Watanabe, N.; Nishi, M.; Ikawa, Y. Requirement of the single base insertion at the 3’ end of the env-related gene of Friend spleen focus-forming virus for pathogenic activity and its effect on localization of the glycoprotein product (gp55). J. Virol. 1989, 63, 4824–4833. [Google Scholar] [CrossRef]
- Srinivas, R.V.; Kilpatrick, D.R.; Tucker, S.; Rui, Z.; Compans, R.W. The hydrophobic membrane-spanning sequences of the gp52 glycoprotein are required for the pathogenicity of Friend spleen focus-forming virus. J. Virol. 1991, 65, 5272–5280. [Google Scholar] [CrossRef]
- Watanabe, N.; Nishi, M.; Ikawa, Y.; Amanuma, H. A deletion in the Friend spleen focus-forming virus env gene is necessary for its product (gp55) to be leukemogenic. J. Virol. 1990, 64, 2678–2686. [Google Scholar] [CrossRef]
- Watanabe, N.; Yugawa, T.; Ikawa, Y.; Amanuma, H. Both the changes of six amino acids and the C-terminal truncation caused by a one-base insertion in the defective env gene of Friend spleen focus-forming virus significantly affect the pathogenic activity of the encoded leukemogenic membrane glycoprotein (gp55). J. Virol. 1995, 69, 7606–7611. [Google Scholar]
- Gliniak, B.C.; Kabat, D. Leukemogenic membrane glycoprotein encoded by Friend spleen focus-forming virus: Transport to cell surfaces and shedding are controlled by disulfide-bonded dimerization and by cleavage of a hydrophobic membrane anchor. J. Virol. 1989, 63, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, S.K.; Linemeyer, D.; Feild, J.; Troxler, D.; Scolnick, E.M. Characterization of a protein found in cells infected with the spleen focus-forming virus that shares immunological cross-reactivity with the gp70 found in mink cell focus-inducing virus particles. J. Virol. 1979, 30, 787–798. [Google Scholar] [CrossRef]
- Ruta, M.; Clarke, S.; Boswell, B.; Kabat, D. Heterogeneous metabolism and subcellular localization of a potentially leukemogenic membrane glycoprotein encoded by Friend erythroleukemia virus. Isolation of viral and cellular processing mutants. J. Biol. Chem. 1982, 257, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, R.V.; Compans, R.W. Membrane association and defective transport of spleen focus-forming virus glycoproteins. J. Biol. Chem. 1983, 258, 14718–14724. [Google Scholar] [CrossRef]
- Ferro, F.E., Jr.; Kozak, S.L.; Hoatlin, M.E.; Kabat, D. Cell surface site for mitogenic interaction of erythropoietin receptors with the membrane glycoprotein encoded by Friend erythroleukemia virus. J. Biol. Chem. 1993, 268, 5741–5747. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Bestwick, R.K.; Spiro, C.; Kabat, D. The membrane glycoprotein of Friend spleen focus-forming virus: Evidence that the cell surface component is required for pathogenesis and that it binds to a receptor. J. Virol. 1987, 61, 2782–2792. [Google Scholar] [CrossRef]
- Ruta, M.; Bestwick, R.; Machida, C.; Kabat, D. Loss of leukemogenicity caused by mutations in the membrane glycoprotein structural gene of Friend spleen focus-forming virus. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 4704–4708. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L.; Mak, T.W.; Bernstein, A. Quantitative colony method for tumorigenic cells transformed by two distinct strains of Friend leukemia virus. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 1703–1707. [Google Scholar] [CrossRef]
- Berger, S.A.; Sanderson, N.; Bernstein, A.; Hankins, W.D. Induction of the early stages of Friend erythroleukemia with helper-free Friend spleen focus-forming virus. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 6913–6917. [Google Scholar] [CrossRef]
- Bestwick, R.K.; Hankins, W.D.; Kabat, D. Roles of helper and defective retroviral genomes in murine erythroleukemia: Studies of spleen focus-forming virus in the absence of helper. J. Virol. 1985, 56, 660–664. [Google Scholar] [CrossRef]
- Wolff, L.; Ruscetti, S. Malignant transformation of erythroid cells in vivo by introduction of a nonreplicating retrovirus vector. Science 1985, 228, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Tambourin, P.; Ruscetti, S. Induction of the autonomous stage of transformation in erythroid cells infected with SFFV: Helper virus is not required. Virology 1986, 152, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Carter, W.A. Friend leukemia: Rapid development of erythropoietin-independent hematopoietic precursors. J. Natl. Cancer Inst. 1975, 54, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.K.; Axelrad, A.A. Erythropoietin-independent erythroid colony formation in vitro by hemopoietic cells of mice infected with friend virus. Int. J. Cancer 1975, 15, 467–482. [Google Scholar] [CrossRef]
- Steinheider, G.; Seidel, H.J.; Kreja, L. Comparison of the biological effects of anemia inducing and polycythemia inducing Friend virus complex. Experientia 1979, 35, 1173–1175. [Google Scholar] [CrossRef]
- Tambourin, P.E.; Wendling, F.; Jasmin, C.; Smadja-Joffe, F. The physiopathology of Friend leukemia. Leuk. Res. 1979, 3, 117–129. [Google Scholar] [CrossRef]
- Hoatlin, M.E.; Kozak, S.L.; Lilly, F.; Chakraborti, A.; Kozak, C.A.; Kabat, D. Activation of erythropoietin receptors by Friend viral gp55 and by erythropoietin and down-modulation by the murine Fv-2r resistance gene. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 9985–9989. [Google Scholar] [CrossRef]
- Li, J.P.; D’Andrea, A.D.; Lodish, H.F.; Baltimore, D. Activation of cell growth by binding of Friend spleen focus-forming virus gp55 glycoprotein to the erythropoietin receptor. Nature 1990, 343, 762–764. [Google Scholar] [CrossRef]
- Ruscetti, S.K.; Janesch, N.J.; Chakraborti, A.; Sawyer, S.T.; Hankins, W.D. Friend spleen focus-forming virus induces factor independence in an erythropoietin-dependent erythroleukemia cell line. J. Virol. 1990, 64, 1057–1062. [Google Scholar] [CrossRef]
- Constantinescu, S.N.; Wu, H.; Liu, X.; Beyer, W.; Fallon, A.; Lodish, H.F. The anemic Friend virus gp55 envelope protein induces erythroid differentiation in fetal liver colony-forming units-erythroid. Blood 1998, 91, 1163–1172. [Google Scholar] [CrossRef]
- Chung, S.W.; Wolff, L.; Ruscetti, S.K. Transmembrane domain of the envelope gene of a polycythemia-inducing retrovirus determines erythropoietin-independent growth. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7957–7960. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Kaminchik, J.; Hankins, W.D.; Ruscetti, S.K. Sequence comparisons of the anemia- and polycythemia-inducing strains of Friend spleen focus-forming virus. J. Virol. 1985, 53, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, S.N.; Liu, X.; Beyer, W.; Fallon, A.; Shekar, S.; Henis, Y.I.; Smith, S.O.; Lodish, H.F. Activation of the erythropoietin receptor by the gp55-P viral envelope protein is determined by a single amino acid in its transmembrane domain. EMBO J. 1999, 18, 3334–3347. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Choi, E.; Nie, L.; Li, J.P. Role of the transmembrane sequence of spleen focus-forming virus gp55 in erythroleukemogenesis. Virology 1998, 252, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.; Jolicoeur, P. Retroviral pathogenesis. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Plainsview, NY, USA, 1997; pp. 475–585. [Google Scholar]
- Lilly, F. Fv-2: Identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J. Natl. Cancer Inst. 1970, 45, 163–169. [Google Scholar] [PubMed]
- Persons, D.A.; Paulson, R.F.; Loyd, M.R.; Herley, M.T.; Bodner, S.M.; Bernstein, A.; Correll, P.H.; Ney, P.A. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat. Genet. 1999, 23, 159–165. [Google Scholar] [CrossRef]
- Friend, C.; Haddad, J.R. Tumor formation with transplants of spleen or liver from mice with virus-induced leukemia. J. Natl. Cancer Inst. 1960, 25, 1279–1285. [Google Scholar]
- Friend, C.; Pateleia, M.C.; DeHarven, E. Erythrocytic maturation in vitro of murine (Friend) virus-induced leukemia cells. Natl. Cancer Inst. Monogr. 1966, 22, 505–520. [Google Scholar]
- Marks, P.A.; Rifkind, R.A. Erythroleukemic differentiation. Annu. Rev. Biochem. 1978, 47, 419–448. [Google Scholar] [CrossRef]
- Friend, C.; Sher, W.; Holland, J.G.; Sato, T. Hemoglobin synthesis in murine virus-induced leukemai cells in vitro: Stimulation of erythroid differentiation by dimethylsulfoxide. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 378–382. [Google Scholar] [CrossRef]
- Richmond, T.D.; Chohan, M.; Barber, D.L. Turning cells red: Signal transduction mediated by erythropoietin. Trends Cell Biol. 2005, 15, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, K.; Hanson, C.; Ohashi, T.; Thompson, D.; Muszynski, K.; Ruscetti, S. Erythroid cells rendered erythropoietin independent by infection with Friend spleen focus-forming virus show constitutive activation of phosphatidylinositol 3-kinase and Akt kinase: Involvement of insulin receptor substrate-related adapter proteins. J. Virol. 2000, 74, 3037–3045. [Google Scholar] [CrossRef] [PubMed]
- Verdier, F.; Chretien, S.; Billat, C.; Gisselbrecht, S.; Lacombe, C.; Mayeux, P. Erythropoietin induces the tyrosine phosphorylation of insulin receptor substrate-2. An alternate pathway for erythropoietin-induced phosphatidylinositol 3-kinase activation. J. Biol. Chem. 1997, 272, 26173–26178. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Sato, K.; Nakajima, H.; McKay, C.; Ney, P.A.; Ihle, J.N. The distal region and receptor tyrosines of the Epo receptor are non-essential for in vivo erythropoiesis. EMBO J. 2001, 20, 3156–3166. [Google Scholar] [CrossRef] [PubMed]
- Muszynski, K.W.; Ohashi, T.; Hanson, C.; Ruscetti, S.K. Both the polycythemia- and anemia-inducing strains of Friend spleen focus-forming virus induce constitutive activation of the Raf-1/mitogen-activated protein kinase signal transduction pathway. J. Virol. 1998, 72, 919–925. [Google Scholar] [CrossRef]
- Muszynski, K.W.; Thompson, D.; Hanson, C.; Lyons, R.; Spadaccini, A.; Ruscetti, S.K. Growth factor-independent proliferation of erythroid cells infected with Friend spleen focus-forming virus is protein kinase C dependent but does not require Ras-GTP. J. Virol. 2000, 74, 8444–8451. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, K.; Hanson, C.; Thompson, D.; Yugawa, T.; Ruscetti, S. Activation of the Jun N-terminal kinase pathway by Friend spleen focus-forming virus and its role in the growth and survival of Friend virus-induced erythroleukemia cells. J. Virol. 2005, 79, 12752–12762. [Google Scholar] [CrossRef]
- Finkelstein, L.D.; Ney, P.A.; Liu, Q.P.; Paulson, R.F.; Correll, P.H. Sf-Stk kinase activity and the Grb2 binding site are required for Epo-independent growth of primary erythroblasts infected with Friend virus. Oncogene 2002, 21, 3562–3570. [Google Scholar] [CrossRef]
- Ohashi, T.; Masuda, M.; Ruscetti, S.K. Induction of sequence-specific DNA-binding factors by erythropoietin and the spleen focus-forming virus. Blood 1995, 85, 1454–1462. [Google Scholar] [CrossRef]
- Ohashi, T.; Masuda, M.; Ruscetti, S.K. Constitutive activation of Stat-related DNA-binding proteins in erythroid cells by the Friend spleen focus-forming virus. Leukemia 1997, 11, 251–254. [Google Scholar]
- Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Schweers, R.L.; Persons, D.A.; Rehg, J.E.; Noguchi, C.T.; Ihle, J.N.; Ney, P.A. Role of erythropoietin receptor signaling in Friend virus-induced erythroblastosis and polycythemia. Blood 2006, 107, 73–78. [Google Scholar] [CrossRef]
- Ruscetti, S.; Hanson, C. National Cancer Institute, Frederick, MD, USA. Unpublished work, 2004.
- Socolovsky, M.; Fallon, A.E.; Wang, S.; Brugnara, C.; Lodish, H.F. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: A direct role for Stat5 in Bcl-XL induction. Cell 1999, 98, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, S.; Yugawa, T. National Cancer Institute, Frederick, MD, USA. Unpublished work, 2005.
- Umehara, D.; Watanabe, S.; Ochi, H.; Anai, Y.; Ahmed, N.; Kannagi, M.; Hanson, C.; Ruscetti, S.; Nishigaki, K. Role of phosphatidylinositol 3-kinase in friend spleen focus-forming virus-induced erythroid disease. J. Virol. 2010, 84, 7675–7682. [Google Scholar] [CrossRef]
- Tsushima, H.; Urata, Y.; Miyazaki, Y.; Fuchigami, K.; Kuriyama, K.; Kondo, T.; Tomonaga, M. Human erythropoietin receptor increases GATA-2 and Bcl-xL by a protein kinase C-dependent pathway in human erythropoietin-dependent cell line AS-E2. Cell Growth Differ. 1997, 8, 1317–1328. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, N.; Lacombe, C.; Muller, O.; Gisselbrecht, S.; Mayeux, P. Multimeric structure of the membrane erythropoietin receptor of murine erythroleukemia cells (Friend cells). Cross-linking of erythropoietin with the spleen focus-forming virus envelope protein. J. Biol. Chem. 1991, 266, 16015–16020. [Google Scholar] [CrossRef]
- Zon, L.I.; Moreau, J.F.; Koo, J.W.; Mathey-Prevot, B.; D’Andrea, A.D. The erythropoietin receptor transmembrane region is necessary for activation by the Friend spleen focus-forming virus gp55 glycoprotein. Mol. Cell. Biol. 1992, 12, 2949–2957. [Google Scholar]
- Constantinescu, S.N.; Keren, T.; Russ, W.P.; Ubarretxena-Belandia, I.; Malka, Y.; Kubatzky, K.F.; Engelman, D.M.; Lodish, H.F.; Henis, Y.I. The erythropoietin receptor transmembrane domain mediates complex formation with viral anemic and polycythemic gp55 proteins. J. Biol. Chem. 2003, 278, 43755–43763. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Subramanian, A.; Hegde, S.; Correll, P.H.; Paulson, R.F. Mutation of the Lyn tyrosine kinase delays the progression of Friend virus induced erythroleukemia without affecting susceptibility. Leuk. Res. 2006, 30, 1141–1149. [Google Scholar] [CrossRef]
- Wang, M.H.; Iwama, A.; Skeel, A.; Suda, T.; Leonard, E.J. The murine stk gene product, a transmembrane protein tyrosine kinase, is a receptor for macrophage-stimulating protein. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3933–3937. [Google Scholar] [CrossRef] [PubMed]
- Waltz, S.E.; Toms, C.L.; McDowell, S.A.; Clay, L.A.; Muraoka, R.S.; Air, E.L.; Sun, W.Y.; Thomas, M.B.; Degen, S.J. Characterization of the mouse Ron/Stk receptor tyrosine kinase gene. Oncogene 1998, 16, 27–42. [Google Scholar] [CrossRef]
- Iwama, A.; Okano, K.; Sudo, T.; Matsuda, Y.; Suda, T. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood 1994, 83, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Rulli, K.; Yugawa, T.; Hanson, C.; Thompson, D.; Ruscetti, S.; Nishigaki, K. Ex vivo and in vivo biological effects of a truncated form of the receptor tyrosine kinase Stk when activated by interaction with the friend spleen focus-forming virus envelope glycoprotein or by point mutation. J. Virol. 2004, 78, 4573–4581. [Google Scholar] [CrossRef]
- Nishigaki, K.; Thompson, D.; Hanson, C.; Yugawa, T.; Ruscetti, S. The envelope glycoprotein of friend spleen focus-forming virus covalently interacts with and constitutively activates a truncated form of the receptor tyrosine kinase Stk. J. Virol. 2001, 75, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ni, S.; Hegde, S.; Wang, X.; Sharda, D.R.; August, A.; Paulson, R.F.; Hankey, P.A. Activation of the N-terminally truncated form of the Stk receptor tyrosine kinase Sf-Stk by Friend virus-encoded gp55 is mediated by cysteine residues in the ecotropic domain of gp55 and the extracellular domain of Sf-Stk. J. Virol. 2010, 84, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Teal, H.E.; Ni, S.; Xu, J.; Finkelstein, L.D.; Cheng, A.M.; Paulson, R.F.; Feng, G.S.; Correll, P.H. GRB2-mediated recruitment of GAB2, but not GAB1, to SF-STK supports the expansion of Friend virus-infected erythroid progenitor cells. Oncogene 2006, 25, 2433–2443. [Google Scholar] [CrossRef]
- Ni, S.; Zhao, C.; Feng, G.S.; Paulson, R.F.; Correll, P.H. A novel Stat3 binding motif in Gab2 mediates transformation of primary hematopoietic cells by the Stk/Ron receptor tyrosine kinase in response to Friend virus infection. Mol. Cell. Biol. 2007, 27, 3708–3715. [Google Scholar] [CrossRef]
- Nishigaki, K.; Hanson, C.; Jelacic, T.; Thompson, D.; Ruscetti, S. Friend spleen focus-forming virus transforms rodent fibroblasts in cooperation with a short form of the receptor tyrosine kinase Stk. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15488–15493. [Google Scholar] [CrossRef]
- Jelacic, T.M.; Thompson, D.; Hanson, C.; Cmarik, J.; Nishigaki, K.; Ruscetti, S. The tyrosine kinase sf-stk and its downstream signals are required for maintenance of friend spleen focus-forming virus-induced fibroblast transformation. J. Virol. 2008, 82, 419–427. [Google Scholar] [CrossRef]
- Hegde, S.; Ni, S.; He, S.; Yoon, D.; Feng, G.S.; Watowich, S.S.; Paulson, R.F.; Hankey, P.A. Stat3 promotes the development of erythroleukemia by inducing Pu.1 expression and inhibiting erythroid differentiation. Oncogene 2009, 28, 3349–3359. [Google Scholar] [CrossRef]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Hegde, S.; Porayette, P.; Yon, M.; Hankey, P.; Paulson, R.F. Friend virus utilizes the BMP4-dependent stress erythropoiesis pathway to induce erythroleukemia. J. Virol. 2008, 82, 382–393. [Google Scholar] [CrossRef]
- Moreau-Gachelin, F.; Tavitian, A.; Tambourin, P. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature 1988, 331, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Schuetze, S.; Kozak, S.L.; Kabat, D. A common site for immortalizing proviral integrations in Friend erythroleukemia: Molecular cloning and characterization. J. Virol. 1989, 63, 4958–4961. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Schuetze, S.; Kozak, S.L.; Kozak, C.A.; Kabat, D. The Sfpi-1 proviral integration site of Friend erythroleukemia encodes the ets-related transcription factor Pu.1. J. Virol. 1991, 65, 464–467. [Google Scholar] [CrossRef]
- Schuetze, S.; Paul, R.; Gliniak, B.C.; Kabat, D. Role of the PU.1 transcription factor in controlling differentiation of Friend erythroleukemia cells. Mol. Cell. Biol. 1992, 12, 2967–2975. [Google Scholar]
- Halupa, A.; Bailey, M.L.; Huang, K.; Iscove, N.N.; Levy, D.E.; Barber, D.L. A novel role for STAT1 in regulating murine erythropoiesis: Deletion of STAT1 results in overall reduction of erythroid progenitors and alters their distribution. Blood 2005, 105, 552–561. [Google Scholar] [CrossRef]
- Nishigaki, K.; Hanson, C.; Ohashi, T.; Spadaccini, A.; Ruscetti, S. Erythroblast transformation by the friend spleen focus-forming virus is associated with a block in erythropoietin-induced STAT1 phosphorylation and DNA binding and correlates with high expression of the hematopoietic phosphatase SHP-1. J. Virol. 2006, 80, 5678–5685. [Google Scholar] [CrossRef]
- Klingmuller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- Fisher, R.C.; Slayton, W.B.; Chien, C.; Guthrie, S.M.; Bray, C.; Scott, E.W. PU.1 supports proliferation of immature erythroid progenitors. Leuk. Res. 2004, 28, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ben David, Y.; Prideaux, V.R.; Chow, V.; Benchimol, S.; Bernstein, A. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene 1988, 3, 179–185. [Google Scholar] [PubMed]
- Lavigueur, A.; Bernstein, A. p53 transgenic mice: Accelerated erythroleukemia induction by Friend virus. Oncogene 1991, 6, 2197–2201. [Google Scholar] [PubMed]
- Mowat, M.; Cheng, A.; Kimura, N.; Bernstein, A.; Benchimol, S. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 1985, 314, 633–636. [Google Scholar] [CrossRef]
- Munroe, D.G.; Peacock, J.W.; Benchimol, S. Inactivation of the cellular p53 gene is a common feature of Friend virus-induced erythroleukemia: Relationship of inactivation to dominant transforming alleles. Mol. Cell. Biol. 1990, 10, 3307–3313. [Google Scholar]
- Prasher, J.M.; Elenitoba-Johnson, K.S.; Kelley, L.L. Loss of p53 tumor suppressor function is required for in vivo progression of Friend erythroleukemia. Oncogene 2001, 20, 2946–2955. [Google Scholar] [CrossRef]
- Schlaberg, R.; Choe, D.J.; Brown, K.R.; Thaker, H.M.; Singh, I.R. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16351–16356. [Google Scholar] [CrossRef]
- Urisman, A.; Molinaro, R.J.; Fischer, N.; Plummer, S.J.; Casey, G.; Klein, E.A.; Malathi, K.; Magi-Galluzzi, C.; Tubbs, R.R.; Ganem, D.; Silverman, R.H.; DeRisi, J.L. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006, 2, e25. [Google Scholar] [CrossRef]
- Lombardi, V.C.; Ruscetti, F.W.; Das Gupta, J.; Pfost, M.A.; Hagen, K.S.; Peterson, D.L.; Ruscetti, S.K.; Bagni, R.K.; Petrow-Sadowski, C.; Gold, B.; Dean, M.; Silverman, R.H.; Mikovits, J.A. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 2009, 326, 585–589. [Google Scholar] [CrossRef]
- Lee, K.; Jones, K.S. The path well traveled: Using mammalian retroviruses to guide research on XMRV. Mol. Interv. 2010, 10, 20–24. [Google Scholar] [CrossRef]
- Kim, S.; Kim, N.; Dong, B.; Boren, D.; Lee, S.A.; Das Gupta, J.; Gaughan, C.; Klein, E.A.; Lee, C.; Silverman, R.H.; Chow, S.A. Integration site preference of xenotropic murine leukemia virus-related virus, a new human retrovirus associated with prostate cancer. J. Virol. 2008, 82, 9964–9977. [Google Scholar] [CrossRef] [PubMed]







© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cmarik, J.; Ruscetti, S. Friend Spleen Focus-Forming Virus Activates the Tyrosine Kinase sf-Stk and the Transcription Factor PU.1 to Cause a Multi-Stage Erythroleukemia in Mice. Viruses 2010, 2, 2235-2257. https://doi.org/10.3390/v2102235
Cmarik J, Ruscetti S. Friend Spleen Focus-Forming Virus Activates the Tyrosine Kinase sf-Stk and the Transcription Factor PU.1 to Cause a Multi-Stage Erythroleukemia in Mice. Viruses. 2010; 2(10):2235-2257. https://doi.org/10.3390/v2102235
Chicago/Turabian StyleCmarik, Joan, and Sandra Ruscetti. 2010. "Friend Spleen Focus-Forming Virus Activates the Tyrosine Kinase sf-Stk and the Transcription Factor PU.1 to Cause a Multi-Stage Erythroleukemia in Mice" Viruses 2, no. 10: 2235-2257. https://doi.org/10.3390/v2102235
APA StyleCmarik, J., & Ruscetti, S. (2010). Friend Spleen Focus-Forming Virus Activates the Tyrosine Kinase sf-Stk and the Transcription Factor PU.1 to Cause a Multi-Stage Erythroleukemia in Mice. Viruses, 2(10), 2235-2257. https://doi.org/10.3390/v2102235