Profound Differences in Virus Population Genetics Correspond to Protection from CD4 Decline Resulting from Feline Lentivirus Coinfection

Abstract
:1. Introduction
2. Results and Discussion
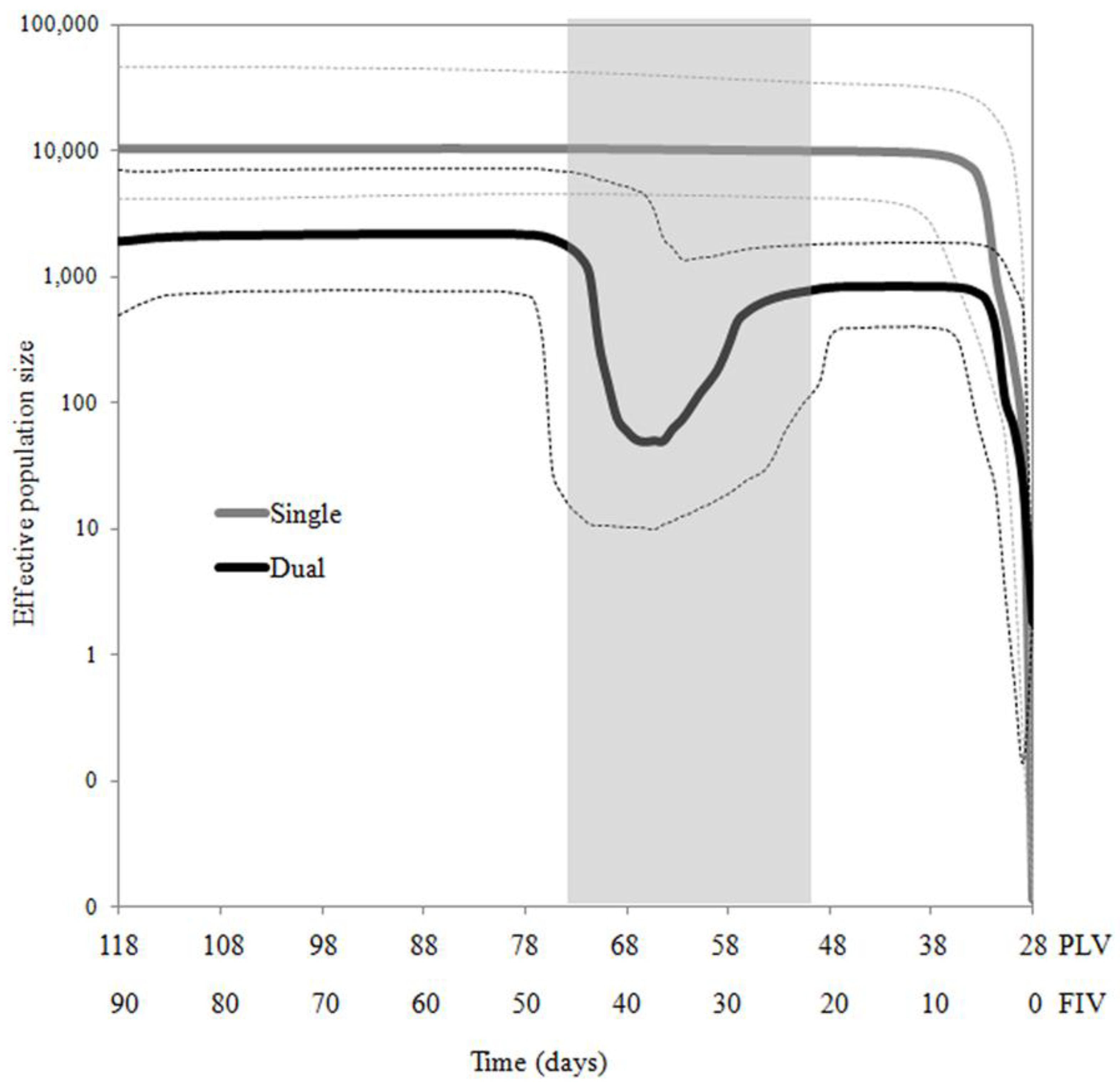
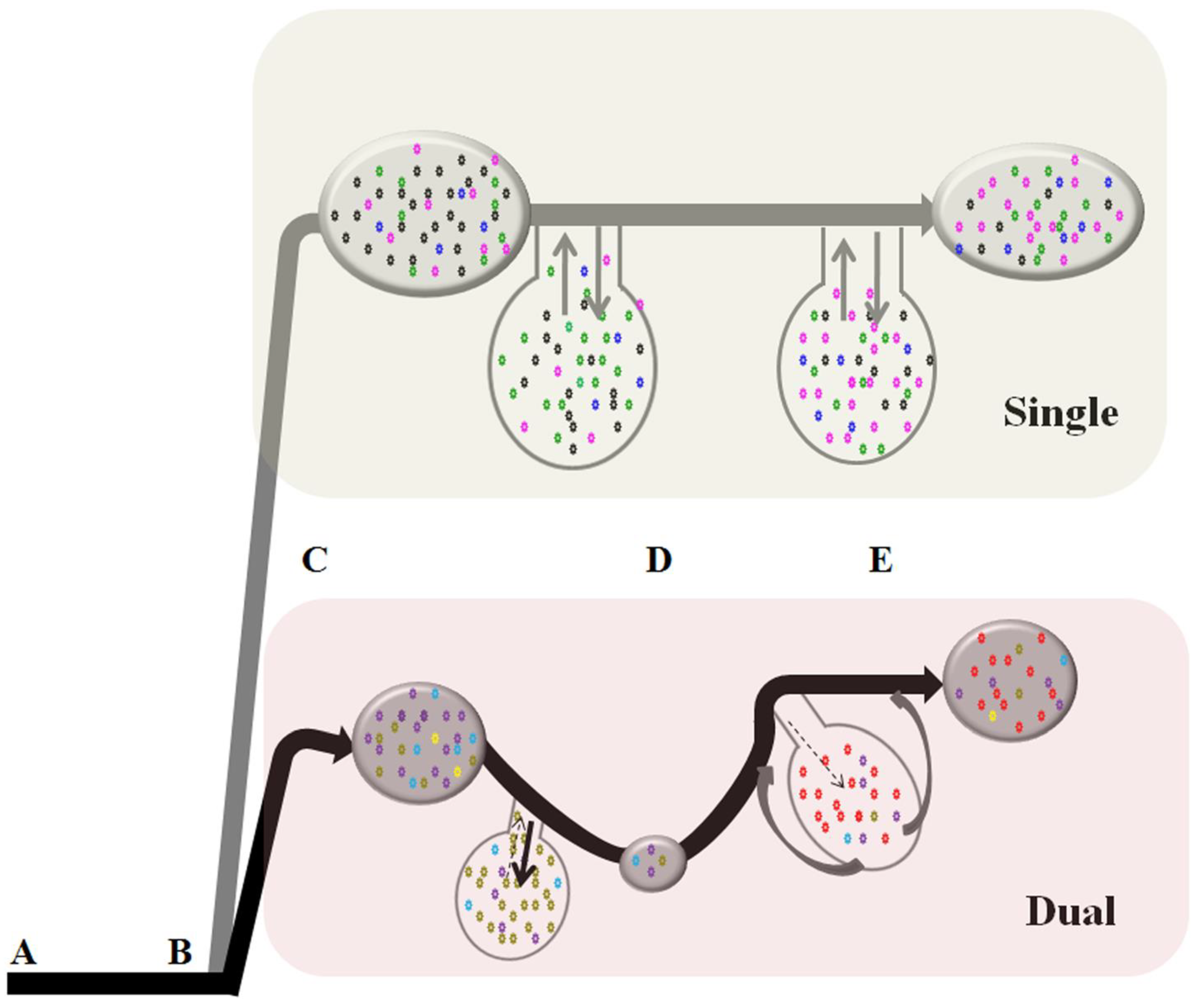
2.1. FIVC Genetic Diversities and Population Dynamics Differ Significantly Between Single and Dual Infection Environments
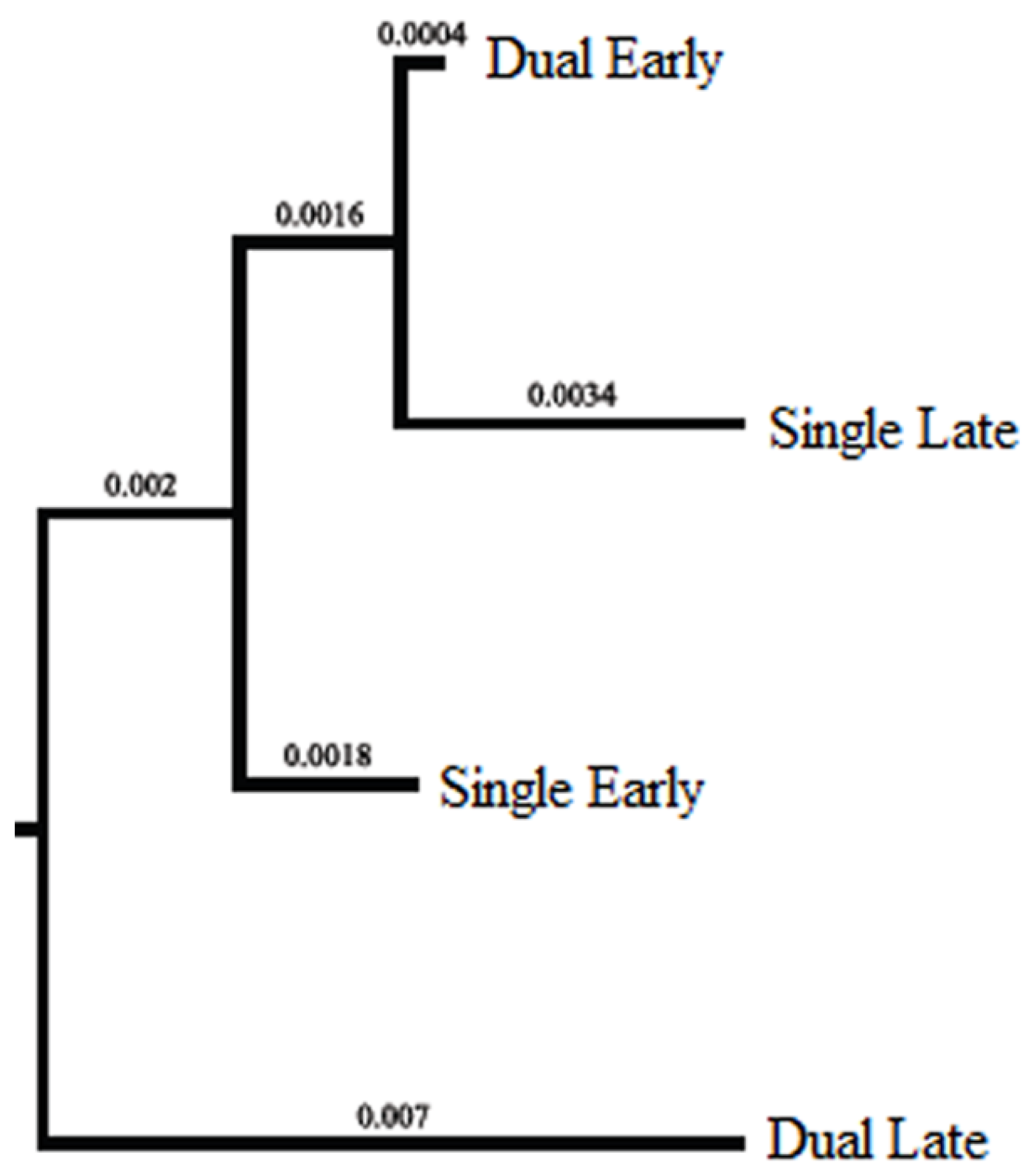
2.2. Temporal Population Structure
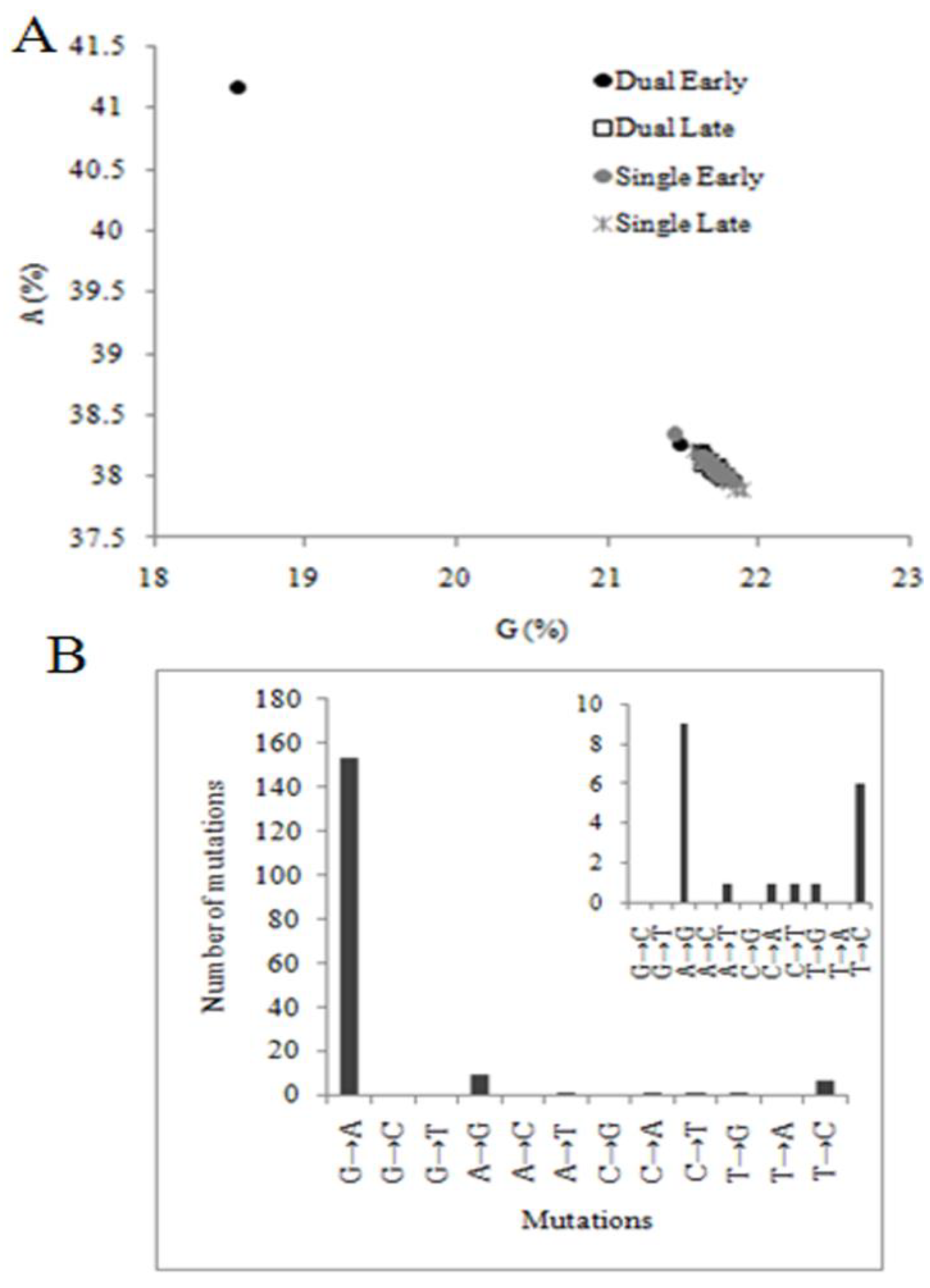
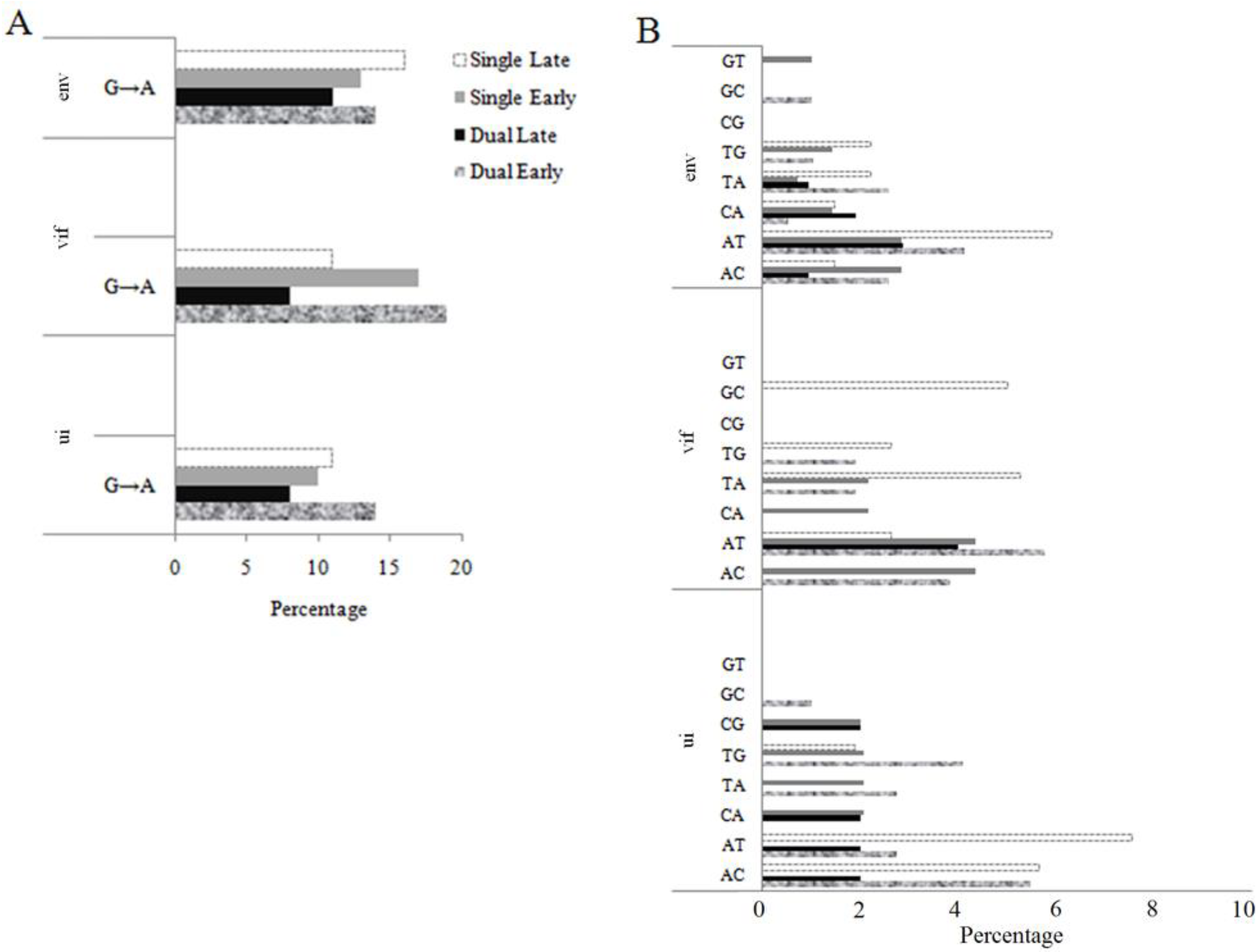
2.3. Pattern of Nucleotide Substitution
2.4. Discussion
3. Materials and methods
3.1. Experimental Design, Cloning, and Sequencing
3.2. Estimates of Genetic Diversities and Population Dynamics
3.3. Analyses of Temporal Population Structure
3.4. Pattern of Nucleotide Substitutions
3.4.1. Selection Analyses
3.4.2. Substitution Bias
Acknowledgements
References and Notes
- Travers, K.; Mboup, S.; Marlink, R.; Guèye-Nidaye, A.; Siby, T.; Thior, I.; Traore, I.; Dieng-Sarr, A.; Sankalé, J.L.; Mullins, C.; Ndoye, I.; Hsieh, C.C.; Essex, M.; Kanki, P. Natural protection against HIV-1 infection provided by HIV-2. Science 1995, 268, 1612–1615. [Google Scholar] [CrossRef]
- Xiang, J.; Wünschmann, S.; Diekema, D.J.; Klinzman, D.; Patrick, K.D.; George, S.L.; Stapleton, J.T. Effect of coinfection with GB virus C on survival among patients with HIV infection. N. Engl. J. Med. 2001, 345, 707–714. [Google Scholar] [CrossRef]
- de Rozieres, S.; Mathiason, C.K.; Rolston, M.R.; Chatterji, U.; Hoover, E.A.; Elder, J.H. Characterization of a highly pathogenic molecular clone of feline immunodeficiency virus clade C. J. Virol. 2004, 78, 8971–8982. [Google Scholar] [CrossRef] [PubMed]
- Terwee, J.A.; Carlson, J.K.; Sprague, W.S.; Sondgeroth, K.S.; Shropshire, S.B.; Troyer, J.L.; VandeWoude, S. Prevention of immunodeficiency virus induced CD4+ T-cell depletion by prior infection with a non-pathogenic virus. Virology 2008, 377, 63–70. [Google Scholar] [CrossRef] [PubMed]
- VandeWoude, S.; Troyer, J.; Poss, M. Restrictions to cross-species transmission of lentiviral infection gleaned from studies of FIV. Vet. Immunol. Immunopathol. 2010, 134, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kuhner, M.K.; Yarnato, J.; Felsenstein, J. Estimating Effective Population Size and Mutation Rate From Sequence Data Using Metropolis-Hastings Sampling. Genetics 1995, 140, 1421–1490. [Google Scholar] [CrossRef]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef]
- Frost, S.D.; Dumaurier, M.J.; Wain-Hobson, S.; Brown, A.J. Genetic drift and within-host metapopulation dynamics of HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 6975–6980. [Google Scholar] [CrossRef]
- Plikat, U.; Nieselt-Struwe, K.; Meyerhans, A. Genetic drift can dominate short-term human immunodeficiency virus type 1 nef quasispecies evolution in vivo. J. Virol. 1997, 71, 4233–4240. [Google Scholar] [CrossRef]
- Shriner, D.; Shankarappa, R.; Jensen, M.A.; Nickle, D.C.; Mittler, J.E.; Margolick, J.B.; Mullins, J.I. Influence of random genetic drift on human immunodeficiency virus type 1 env evolution during chronic infection. Genetics 2004, 166, 1155–1164. [Google Scholar] [CrossRef]
- Voronin, Y.; Holte, S.; Overbaugh, J.; Emerman, M. Genetic drift of HIV populations in culture. PLoS Genet. 2009, 5, e1000431. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. 1964, 1, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Li, H.; Schneider, W.L.; Sherman, D.J.; Gray, S.; Smith, D.; Roossinck, M.J. Analysis of genetic bottlenecks during horizontal transmission of Cucumber mosaic virus. J. Virol. 2006, 80, 8345–8350. [Google Scholar] [CrossRef] [PubMed]
- Chao, L. Fitness of RNA virus decreased by Muller’s ratchet. Nature 1990, 348, 454–455. [Google Scholar] [CrossRef]
- Clarke, D.K.; Duarte, E.A.; Moya, A.; Elena, S.F.; Domingo, E.; Holland, J. Genetic bottlenecks and population passages cause profound fitness differences in RNA viruses. J. Virol. 1993, 67, 222–228. [Google Scholar] [CrossRef]
- Duarte, E.; Clarke, D.; Moya, A.; Domingo, E.; Holland, J. Rapid fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 6015–6019. [Google Scholar] [CrossRef]
- Li, H.; Roossinck, M.J. Genetic bottlenecks reduce population variation in an experimental RNA virus population. J. Virol. 2004, 78, 10582–10587. [Google Scholar] [CrossRef]
- Novella, I.S.; Duarte, E.A.; Elena, S.F.; Moya, A.; Domingo, E.; Holland, J.J. Exponential increases of RNA virus fitness during large population transmissions. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 5841–5844. [Google Scholar] [CrossRef]
- van Opijnen, T.; de Ronde, A.; Boerlijst, M.C.; Berkhout, B. Adaptation of HIV-1 depends on the host-cell environment. PLoS One 2007, 2, e271. [Google Scholar] [CrossRef]
- Yuste, E.; López-Galíndez, C.; Domingo, E. Unusual distribution of mutations associated with serial bottleneck passages of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9546–9552. [Google Scholar] [CrossRef]
- Yuste, E.; Sánchez-Palomino, S.; Casado, C.; Domingo, E.; López-Galíndez, C. Drastic fitness loss in human immunodeficiency virus type 1 upon serial bottleneck events. J. Virol. 1999, 73, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Poss, M.; Ross, H.A.; Painter, S.L.; Holley, D.C.; Terwee, J.A.; Vandewoude, S.; Rodrigo, A. Feline lentivirus evolution in cross-species infection reveals extensive G-to-A mutation and selection on key residues in the viral polymerase. J. Virol. 2006, 80, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Kijak, G.H.; Janini, M.; Tovanabutra, S.; Sanders-Buell, E.E.; Birx, D.L.; Robb, M.L.; Michael, N.L.; McCutchan, F.E. HyperPack: A software package for the study of levels, contexts, and patterns of APOBEC-mediated hypermutation in HIV. AIDS Res. Hum. Retrovirus. 2007, 23, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Becquart, P.; Chomont, N.; Roques, P.; Ayouba, A.; Kazatchkine, M.D.; Belec, L.; Hocini, H. Compartmentalization of HIV-1 between breast milk and blood of HIV-infected mothers. Virology 2002, 300, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Bull, M.; Learn, G.; Genowati, I.; McKernan, J.; Hitti, J.; Lockhart, D.; Tapia, K.; Holte, S.; Dragavon, J.; Coombs, R.; Mullins, J.; Frenkel, L. Compartmentalization of HIV-1 within the female genital tract is due to monotypic and low-diversity variants not distinct viral populations. PLoS One 2009, 4, e7122. [Google Scholar] [CrossRef]
- Delwart, E.L.; Mullins, J.I.; Gupta, P.; Learn, Jr., G.H.; Holodniy, M.; Katzenstein, D.; Walker, B.D.; Singh, M.K. Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 1998, 72, 617–623. [Google Scholar] [CrossRef]
- Hayward, J.J.; Rodrigo, A.G. The distribution of feline immunodeficiency virus in tissue compartments of feral domestic cats. Arch. Virol. 2010, 155, 411–416. [Google Scholar] [CrossRef]
- Kiessling, A.A.; Fitzgerald, L.M.; Zhang, D.; Chhay, H.; Brettler, D.; Eyre, R.C.; Steinberg, J.; McGowan, K.; Byrn, R.A. Human immunodeficiency virus in semen arises from a genetically distinct virus reservoir. AIDS Res. Hum. Retrovirus. 1998, 14, S33–S41. [Google Scholar]
- Marras, D.; Bruggeman, L.A.; Gao, F.; Tanji, N.; Mansukhani, M.M.; Cara, A.; Ross, M.D.; Gusella, G.L.; Benson, G.; D’Agati, V.D.; Hahn, B.H.; Klotman, M.E.; Klotman, P.E. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat. Med. 2002, 8, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Poss, M.; Rodrigo, A.G.; Gosink, J.J.; Learn, G.H.; deVange, P.D.; Martin Jr., H.L.; Bwayo, J.; Kreiss, J.K.; Overbaugh, J. Evolution of envelope sequences from the genital tract and peripheral blood of women infected with clade A human immunodeficiency virus type 1. J. Virol. 1998, 72, 8240–8251. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997a, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Ignacio, C.C.; Torriani, F.; Havlir, D.; Fitch, N.J.; Richman, D.D. In vivo compartmentalization of human immunodeficiency virus: Evidence from the examination of pol sequences from autopsy tissues. J. Virol. 1997b, 71, 2059–2071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rowe, L.; He, T.; Chung, C.; Yu, J.; Yu, W.; Talal, A.; Markowitz, M.; Ho, D.D. Compartmentalization of surface envelope glycoprotein of human immunodeficiency virus type 1 during acute and chronic infection. J. Virol. 2002, 76, 9465–9473. [Google Scholar] [CrossRef]
- Reeve, A.B.; Patel, K.; Pearce, N.C.; Augustus, K.V.; Domingues, H.G.; O’Neil, S.P.; Novembre, F.J. Reduced genetic diversity in lymphoid and central nervous system tissues and selection-induced tissue-specific compartmentalization of neuropathogenic SIVsmmFGb during acute infection. AIDS Res. Hum. Retrovirus. 2009, 6, 583–601. [Google Scholar] [CrossRef]
- Schnell, G.; Price, R.W.; Swanstrom, R.; Spudich, S. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J. Virol. 2010, 84, 2395–2407. [Google Scholar] [CrossRef]
- Roy, S.; Lavine, J.; Chiaromonte, F.; Terwee, J.; VandeWoude, S.; Bjornstad, O.; Poss, M. Multivariate statistical analyses demonstrate unique host immune responses to single and dual lentiviral infection. PLoS One 2009, 4, e7359. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, H.; Mansky, L.M. Roles of uracil-DNA glycosylase and dUTPase in virus replication. J. Gen. Virol. 2002, 83, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Yu, Q.; Landau, N.R. New insights into the role of Vif in HIV-1 replication. AIDS Rev. 2004, 6, 34–39. [Google Scholar] [PubMed]
- Koning, F.A.; Newman, E.N.C.; Kim, E.U.; Kunstman, K.J.; Steven, M.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 Expression Patterns in Human Tissues and Hematopoietic Cell Subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed]
- Lerner, D.L.; Wagaman, P.C.; Phillips, T.R.; Prospero-Garcia, O.; Henriksen, S.J.; Fox, H.S.; Bloom, F.E.; Elder, J.H. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 7480–7484. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Watterson, G.A. On the number of segregating sites in genetical systems without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNApolymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v1.4. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 24 November 2010).
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin version 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.; Quattro, J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.69; Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2009. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G→A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods), version 4.0b10; Sinauer Associates: Sunderland, Massachusetts, MA, USA, 2002. [Google Scholar]
- Rambaut, A.; Grassly, N. Seq-Gen: An application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 1997, 13, 235–238. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Clock model | Mutation rate (×10−5/site/day) | CoV* |
|---|---|---|---|
| Single | Strict | 10.60 (9.23–12.5) | N.A |
| Relaxed | 4.60 (4.21–5.00) | 0.866 (0.698–1.057) | |
| Dual | Strict | 11.3 (9.87–12.9) | N.A |
| Relaxed | 6.94 (5.27–8.12) | 0.884 (0.681–1.116) |
| Source of | Sum of | Variance | Percentage | Fixation | |
|---|---|---|---|---|---|
| variation | d.f. | squares | components | of variation | index |
| Among | |||||
| groups | 1 | 11.208 | −0.01690 Va | −0.14 | FCT: −0.00175 |
| Among | |||||
| populations | |||||
| within groups | 2 | 24.364 | 0.08847 Vb | 0.92 | *FSC: 0.00915 |
| Within | |||||
| populations | 114 | 1092.68 | 9.58492 Vc | 99.26 | *FST: 0.00741 |
| Total | 117 | 1128.25 | 9.65649 |
| ui | vif | env | ||||||
|---|---|---|---|---|---|---|---|---|
| Kappa (k = Ts/Tv) | Early | Late | Early | Late | Early | Late | ||
| Single | 17.1712 | 10.2651 | 9.0782 | 7.88854 | 14.8736 | 7.88854 | ||
| Dual | 8.868 | 20.1281 | 10.2285 | 33.0202 | 13.6558 | 26.5212 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padhi, A.; Ross, H.; Terwee, J.; VandeWoude, S.; Poss, M. Profound Differences in Virus Population Genetics Correspond to Protection from CD4 Decline Resulting from Feline Lentivirus Coinfection. Viruses 2010, 2, 2663-2680. https://doi.org/10.3390/v2122663
Padhi A, Ross H, Terwee J, VandeWoude S, Poss M. Profound Differences in Virus Population Genetics Correspond to Protection from CD4 Decline Resulting from Feline Lentivirus Coinfection. Viruses. 2010; 2(12):2663-2680. https://doi.org/10.3390/v2122663
Chicago/Turabian StylePadhi, Abinash, Howard Ross, Julie Terwee, Sue VandeWoude, and Mary Poss. 2010. "Profound Differences in Virus Population Genetics Correspond to Protection from CD4 Decline Resulting from Feline Lentivirus Coinfection" Viruses 2, no. 12: 2663-2680. https://doi.org/10.3390/v2122663
APA StylePadhi, A., Ross, H., Terwee, J., VandeWoude, S., & Poss, M. (2010). Profound Differences in Virus Population Genetics Correspond to Protection from CD4 Decline Resulting from Feline Lentivirus Coinfection. Viruses, 2(12), 2663-2680. https://doi.org/10.3390/v2122663