Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics


Abstract
:1. Ectromelia virus
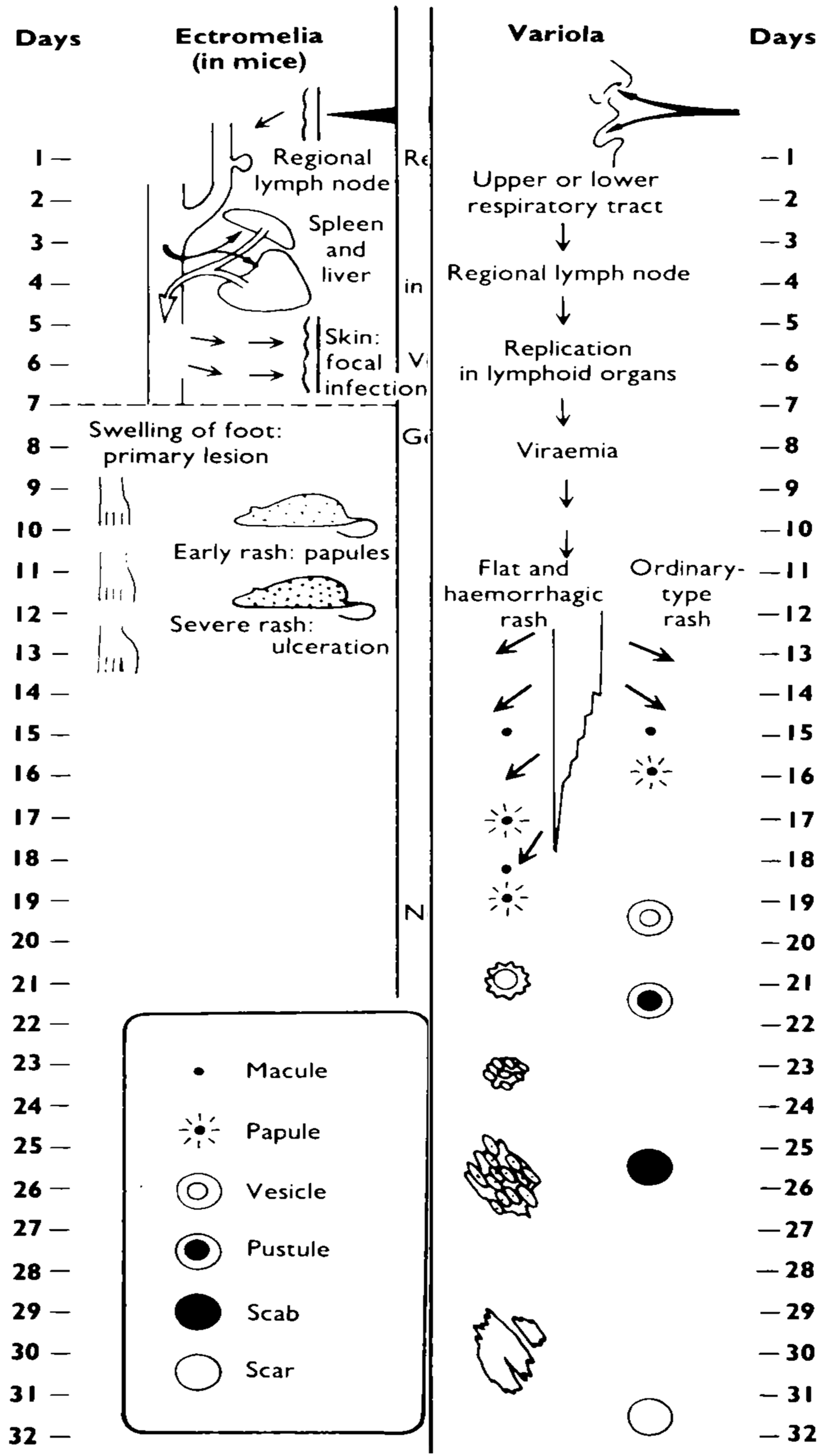
Mousepox as a model of smallpox
2. The animal efficacy rule
3. Mousepox severity is dependent on mouse strain
4. Importance of route and infectious dose in animal models of smallpox
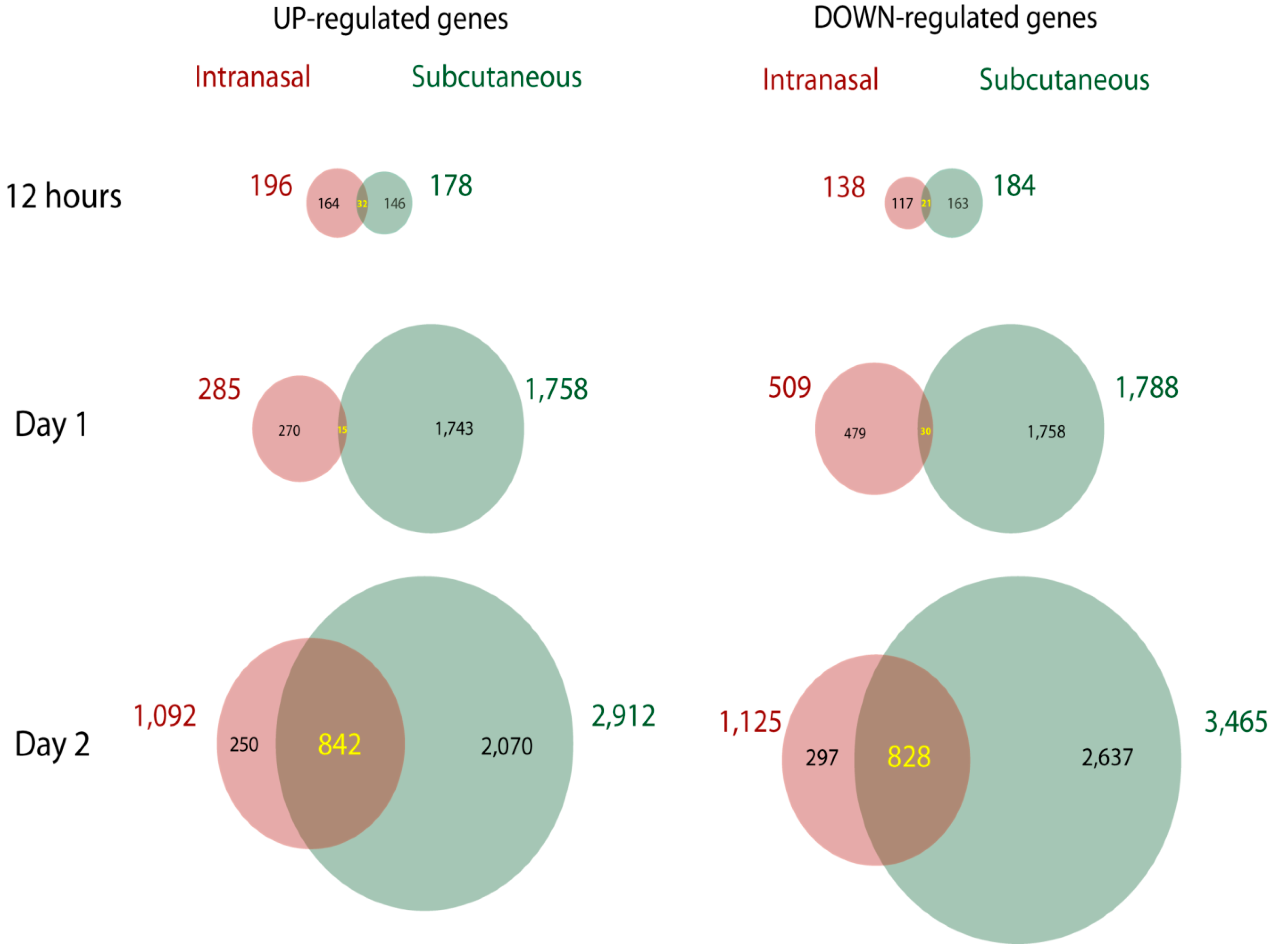
4.1. Route of infection
4.2. Infectious Dose
5. Indicators of disease progression and recovery from infection
5.1. Disease progression
5.2. Recovery from infection
6. Selecting a trigger for therapeutic intervention

{kind=link}
{kind=link}
| Marker | Route | A/Ncr | C57BL/6 |
| Disease Progression | |||
| Day of death | IN | 7-8 (1000 PFU); 7-12 (20 PFU) | 9-14 (1000 PFU); |
| FP | 7-8 (1000 PFU) | N/A | |
| Weight change | IN | Lose weight from day 5 (5-1000 PFU) | Lose weight from day 7 (1000 PFU) |
| FP | Lose weight from day 5 (1000 PFU) | N/A | |
| Infectivity titres | IN | Day 2 spleen, liver and lung (1000 PFU) | (Day 3 liver; day 4 spleen and lung 1000 PFU) |
| FP | Day 2 spleen; day 4 liver; and day 6 lung (1000 PFU) | N/A | |
| ALT/AST | IN | Day 6 (1500 PFU) | >Day 7 (1500 PFU); day 5 (1x106 PFU) |
| Blood viral DNA | IN | Day 6 (5 PFU) | Day 4 (6500 PFU) |
| FP | Day 5 (1000 PFU) | - | |
| Host Response | |||
| IFN-γ | IN | Day 4 (20 PFU) | Day 4 (1x106 PFU) |
| SC | - | Day 2 (1x106 PFU) | |
| Neutrophilia | IN | By day 8 (20 PFU) | By day 6 (1x106 PFU) |
| PLN IFN-γ | FP | Day 2 (1000 PFU) | Day 1 (1000 PFU) |
| PLN Rantes | FP | No change (1000 PFU) | Day 1 (1000 PFU) |
| PLN IL-9 | FP | Day 1 (1000 PFU) | No change (1000 PFU) |
| PLN gene regulation | FP | 22 gene changes from 6-24 hours p.i. (1000 PFU) | 80 gene changes from 6-24 hours p.i. (1000 PFU) |
| Antigen presentation | IN | MLN no presentation up to day 3 (1x106 PFU) | MLN no presentation up to day 3 (1x106 PFU) |
| FP | PLN no presentation up to day 3 (1x106 PFU) | PLN presentation from day 1 (1x106 PFU) | |
| Spleen mass | IN | No change | Doubled from day 4-7 |
| FP | No change | Trebled from day 4-7 | |
| CD4 splenic intracellular IFN-γ | FP | 2x105 IFN- γ + cells by day 8 (3000 PFU attenuated virus EV-138) | 5x104 IFN-γ + cells by day 6 (3000 PFU attenuated virus EV-138) |
| CD8 splenic intracellular IFN-γ | FP | 2.5x105 IFN- γ + cells by day 8 (3000 PFU attenuated virus EV-138) | 2.5x106 IFN-γ+ cells by day 6 (3000 PFU attenuated virus EV-138) |
| Antibody | FP | N/A (1000 PFU) | Seroconversion by day 21 |
7. Evaluation of prophylactics and therapeutics in the ectromelia model
7.1. Efficacy testing in ECTV infected immunocompetent mice
7.2. Efficacy testing smallpox antivirals in immunodeficient animals
References and Notes
- Marchal, J. Infectious ectromelia. A hitherto undescribed virus disease in mice. J. Pathol. Bacteriol. 1930, 33, 713–728. [Google Scholar] [CrossRef]
- Fenner, F. Mousepox (infectious ectromelia): past, present, and future. Lab. Anim. Sci. 1981, 31, 553–559. [Google Scholar] [PubMed]
- Buller, R.M.; Palumbo, G.J. Poxvirus pathogenesis. Microbiol.Rev. 1991, 55, 80–122. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.L.; Fenner, F. The mouse in biomedical research; Elsevier: New York, NY, USA, 2007; pp. 67–92. [Google Scholar]
- Dick, E.J.; JrKittell, C.L.; Meyer, H.; Farrar, P.L.; Ropp, S.L.; Esposito, J.J. Mousepox outbreak in laboratory mouse colony. Lab. Anim. Sci. 1996, 46, 602–611. [Google Scholar]
- Buller, R.M.; Potter, M.; Wallace, G.D. Variable resistance to ectromelia (mousepox) virus among genera of Mus. Curr. Top. Microbiol. Immunol. 1986, 127, 319–322. [Google Scholar]
- Parker, S.; Handley, L.; Buller, R.M. Therapeutic and prophylactic drugs to treat orthopoxvirus infections. Future Virology 2008, 3, 595–612. [Google Scholar] [CrossRef]
- Fenner, F. The pathogenesis of the acute exanthems; an interpretation based on experimental investigations with mousepox; infectious ectromelia of mice. Lancet 1948, 2, 915–920. [Google Scholar] [CrossRef]
- Esteban, D.J.; Buller, R.M. Ectromelia virus: the causative agent of mousepox. J. Gen. Virol. 2005, 86, 2645–2659. [Google Scholar] [CrossRef]
- Chaudhri, G.; Panchanathan, V.; Buller, R.M.; van den Eertwegh, A.J.; Claassen, E.; Zhou, J.; de Chazal, R.; Laman, J.D.; Karupiah, G. Polarized type 1 cytokine response and cell-mediated immunity determine genetic resistance to mousepox. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9057–9062. [Google Scholar] [CrossRef]
- Fang, M.; Lanier, L.L.; Sigal, L.J. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. 2008, 4, e30. [Google Scholar] [CrossRef]
- Fang, M.; Sigal, L. Studying NK cell responses to ectromelia virus infections in mice. Methods Mol. Biol. 2010, 612, 411–428. [Google Scholar] [PubMed]
- Fang, M.; Sigal, L.J. Antibodies and CD8+ T cells are complementary and essential for natural resistance to a highly lethal cytopathic virus. J. Immunol. 2005, 175, 6829–6836. [Google Scholar] [CrossRef] [PubMed]
- Karupiah, G. Type 1 and type 2 cytokines in antiviral defense. Vet. Immunol. Immunopathol. 1998, 63, 105–109. [Google Scholar] [CrossRef]
- Panchanathan, V.; Chaudhri, G.; Karupiah, G. Protective immunity against secondary poxvirus infection is dependent on antibody but not on CD4 or CD8 T-cell function. J. Virol. 2006, 80, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.K.; Parker, S.; Yokoyama, W.M.; Corbett, J.A.; Buller, R.M. Induction of natural killer cell responses by ectromelia virus controls infection. J. Virol. 2007, 81, 4070–4079. [Google Scholar] [CrossRef]
- Parker, A.K.; Yokoyama, W.M.; Corbett, J.A.; Chen, N.; Buller, R.M. Primary naive and interleukin-2-activated natural killer cells do not support efficient ectromelia virus replication. J. Gen. Virol. 2008, 89, 751–759. [Google Scholar] [CrossRef]
- Tscharke, D.C.; Woo, W.P.; Sakala, I.G.; Sidney, J.; Sette, A.; Moss, D.J.; Bennink, J.R.; Karupiah, G.; Yewdell, J.W. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J. Virol. 2006, 80, 6318–6323. [Google Scholar] [CrossRef]
- Wang, Y.; Chaudhri, G.; Jackson, R.J.; Karupiah, G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J. Immunol. 2009, 183, 3324–3331. [Google Scholar] [CrossRef]
- Brownstein, D.G. Comparative genetics of resistance to viruses. Am. J. Hum. Genet. 1998, 62, 211–214. [Google Scholar] [CrossRef]
- Brownstein, D.G.; Bhatt, P.N.; Gras, L.; Budris, T. Serial backcross analysis of genetic resistance to mousepox, using marker loci for Rmp-2 and Rmp-3. J. Virol. 1992, 66, 7073–7079. [Google Scholar] [CrossRef]
- Brownstein, D.G.; Gras, L. Chromosome mapping of Rmp-4, a gonad-dependent gene encoding host resistance to mousepox. J. Virol. 1995, 69, 6958–6964. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.L.; Brownstein, D.G. Innate resistance to lethal mousepox is genetically linked to the NK gene complex on chromosome 6 and correlates with early restriction of virus replication by cells with an NK phenotype. J. Virol. 1995, 69, 5875–5877. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Smallpox and its eradication; World Health Organisation: Geneva, Switzerland, 1988. [Google Scholar]
- Roberts, J.A. Histopathogenesis of mousepox. I. Respiratory infection. Br. J. Exp. Pathol. 1962, 43, 451–461. [Google Scholar] [PubMed]
- Parker, S.; Nuara, A.; Buller, R.M.; Schultz, D.A. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2007, 2, 17–34. [Google Scholar] [CrossRef]
- Jordan, R.; Hruby, D. Smallpox antiviral drug development: satisfying the animal efficacy rule. Expert Rev. Anti. Infect. Ther. 2006, 4, 277–289. [Google Scholar] [CrossRef]
- Painter, G.; Buller, R.M.L.; Huggins, J.; Moyer, R.W.; Painter, W.; Doucette, M. The challenge of developing an antiviral agent for the treatment of smallpox using the animal efficacy rule. Future Virology 2006, 1, 173–179. [Google Scholar] [CrossRef]
- Parker, S.; Siddiqui, A.M.; Oberle, C.; Hembrador, E.; Lanier, R.; Painter, G.; Robertson, A.; Buller, R.M. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 2009, 385, 11–21. [Google Scholar] [CrossRef]
- Jacoby, R.O.; Bhatt, P.N. Mousepox in inbred mice innately resistant or susceptible to lethal infection with ectromelia virus. II. Pathogenesis. Lab. Anim. Sci. 1987, 37, 16–22. [Google Scholar]
- Parker, S.; Buller, R.M. Mousepox disease is dependent on route of infection. Saint Louis University: St Louis, MO, USA, 2010. [Google Scholar]
- Hooper, J.W.; Thompson, E.; Wilhelmsen, C.; Zimmerman, M.; Ichou, M.A.; Steffen, S.E.; Schmaljohn, C.S.; Schmaljohn, A.L.; Jahrling, P.B. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 2004, 78, 4433–4443. [Google Scholar] [CrossRef]
- Adams, M.M.; Rice, A.D.; Moyer, R.W. Rabbitpox virus and vaccinia virus infection of rabbits as a model for human smallpox. J. Virol. 2007, 81, 11084–11095. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Hensley, L.E.; Martinez, M.J.; Leduc, J.W.; Rubins, K.H.; Relman, D.A.; Huggins, J.W. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15196–15200. [Google Scholar] [CrossRef] [PubMed]
- Mims, C.A. The response of mice to large intravenous injections of ectromelia virus. II. The growth of virus in the liver. Br. J. Exp. Pathol. 1959, 40, 543–550. [Google Scholar] [PubMed]
- Moulton, E.A.; Bertram, P.; Chen, N.H.; Buller, R.M.L.; Atkinson, J.P. The interaction between the host’s complement system and the poxviral complement regulatory protein determines the lethality of ectromelia infection. Molecular Immunology 2008, 45, 4113–4113. [Google Scholar] [CrossRef]
- Meiklejohn, G.K.; Kempe, C.H.; Downie, A.W.; Berge, T.O.; St. Vincent, L.; Rao, A.R. Air sampling to recover variola virus in the environment of a smallpox hospital. Bull. World Health Organisation 1961, 25, 63–67. [Google Scholar]
- Downie, A.W.; Meiklejohn, M.; St. Vincent, L.; Rao, A.R.; Sundara, B.V.; Kempe, C.H. The recovery of smallpox virus from patients and their environment in a smallpox hospital. Bull. World Health Organisation 1965, 33, 615–622. [Google Scholar]
- Zelicoff, A.P. An epidemiological analysis of the 1971 smallpox outbreak in Aralsk, Kazakhstan. Crit Rev. Microbiol. 2003, 29, 97–108. [Google Scholar] [CrossRef]
- Parker, S.; Schriewer, J.; Oberle, C.; Robertson, A.; Lanier, R.; Painter, G.; Buller, R.M. Using biomarkers to stage disease progression in a lethal mousepox model treated with CMX001. Antivir. Ther. 2008, 13, 863–873. [Google Scholar] [CrossRef]
- Gronvall, G.K.; Trent, D.; Borio, L.; Brey, R.; Nagao, L. The FDA animal efficacy rule and biodefense. Nat. Biotechnol. 2007, 25, 1084–1087. [Google Scholar]
- Parker, S.; Touchette, E.; Oberle, C.; Almond, M.; Robertson, A.; Trost, L.C.; Lampert, B.; Painter, G.; Buller, R.M. Efficacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res. 2008, 77, 39–49. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Buller, R.M.; Parker, S.; Keith, K.A.; Hruby, D.E.; Jordan, R.; Kern, E.R. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob.Agents Chemother. 2007, 51, 689–695. [Google Scholar] [CrossRef]
- Vora, S.; Damon, I.; Fulginiti, V.; Weber, S.G.; Kahana, M.; Stein, S.L.; Gerber, S.I.; Garcia-Houchins, S.; Lederman, E.; Hruby, D.; Collins, L.; Scott, D.; Thompson, K.; Barson, J.V.; Regnery, R.; Hughes, C.; Daum, R.S.; Li, Y.; Zhao, H.; Smith, S.; Braden, Z.; Karem, K.; Olson, V.; Davidson, W.; Trindade, G.; Bolken, T.; Jordan, R.; Tien, D.; Marcinak, J. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin. Infect. Dis. 2008, 46, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- CDC, Progressive vaccinia in a military smallpox vacinee - United States. MMWR Morb. Mortal. Wkly. Rep. 2009, 58, 1–4.
- Bray, M.; Martinez, M.; Smee, D.F.; Kefauver, D.; Thompson, E.; Huggins, J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infec. Dis. 2000, 181, 10–19. [Google Scholar] [CrossRef]
- Neyts, J.; Leyssen, P.; Verbeken, E.; De Clercq, E. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob Agents Chemother. 2004, 48, 2267–2273. [Google Scholar] [CrossRef]
- Smee, D.F.; Bailey, K.W.; Wong, M.H.; Wandersee, M.K.; Sidwell, R.W. Topical cidofovir is more effective than is parenteral therapy for treatment of progressive vaccinia in immunocompromised mice. J. Infect. Dis. 2004, 190, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Grosenbach, D.W.; Berhanu, A.; King, D.S.; Mosier, S.; Jones, K.F.; Jordan, R.A.; Bolken, T.C.; Hruby, D.E. Efficacy of ST-246 versus lethal poxvirus challenge in immunodeficient mice. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 838–843. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Prichard, M.N.; Keith, K.A.; Hruby, D.E.; Jordan, R.; Painter, G.R.; Robertson, A.; Kern, E.R. Synergistic efficacy of the combination of ST-246 with CMX001 against orthopoxviruses. Antimicrob. Agents Chemother. 2007, 51, 4118–4124. [Google Scholar] [CrossRef] [PubMed]
- Stabenow, J.; Buller, R.M.; Schriewer, J.; West, C.; Sagartz, J.E.; Parker, S. A mouse model of lethal infection for evaluating prophylactics and therapeutics against Monkeypox virus. J. Virol. 2010, 84, 3909–3920. [Google Scholar] [CrossRef] [PubMed]
- Handley, L.; Buller, R.M.; Frey, S.E.; Bellone, C.; Parker, S. The new ACAM2000 vaccine and other therapies to control orthopoxvirus outbreaks and bioterror attacks. Expert Rev. Vaccines 2009, 8, 841–850. [Google Scholar] [CrossRef]
- Jackson, R.J.; Ramsay, A.J.; Christensen, C.D.; Beaton, S.; Hall, D.F.; Ramshaw, I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J Virol. 2001, 75, 1205–1210. [Google Scholar] [CrossRef]


| Criteria for use of animal model | Issues relating to smallpox |
|---|---|
| There is reasonably well understood pathophysiological mechanism for the toxicity of the substance and its prevention or substantial reduction by the product. |
|
| The effect is demonstrated in more than one animal species expected to react with a response predictive for humans, unless the effect is demonstrated in a single animal species that represents a sufficiently well characterized animal model for predicting human response. |
|
| The animal study endpoint is related clearly to the desired benefit in humans, generally the enhancement of survival or prevention of major morbidity. |
|
| The data or information on the pharmacokinetics and pharmacodynamics of the product or other relevant data or information, in animals and humans, allow selection of an effective human dose. |
|
© 2010 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Parker, S.; Siddiqui, A.M.; Painter, G.; Schriewer, J.; Buller, R.M. Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics. Viruses 2010, 2, 1918-1932. https://doi.org/10.3390/v2091918
Parker S, Siddiqui AM, Painter G, Schriewer J, Buller RM. Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics. Viruses. 2010; 2(9):1918-1932. https://doi.org/10.3390/v2091918
Chicago/Turabian StyleParker, Scott, Akbar M. Siddiqui, George Painter, Jill Schriewer, and R. Mark Buller. 2010. "Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics" Viruses 2, no. 9: 1918-1932. https://doi.org/10.3390/v2091918
APA StyleParker, S., Siddiqui, A. M., Painter, G., Schriewer, J., & Buller, R. M. (2010). Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics. Viruses, 2(9), 1918-1932. https://doi.org/10.3390/v2091918