N-Terminally Myristoylated Feline Foamy Virus Gag Allows Env-Independent Budding of Sub-Viral Particles

Abstract
:1. Introduction
2. Results
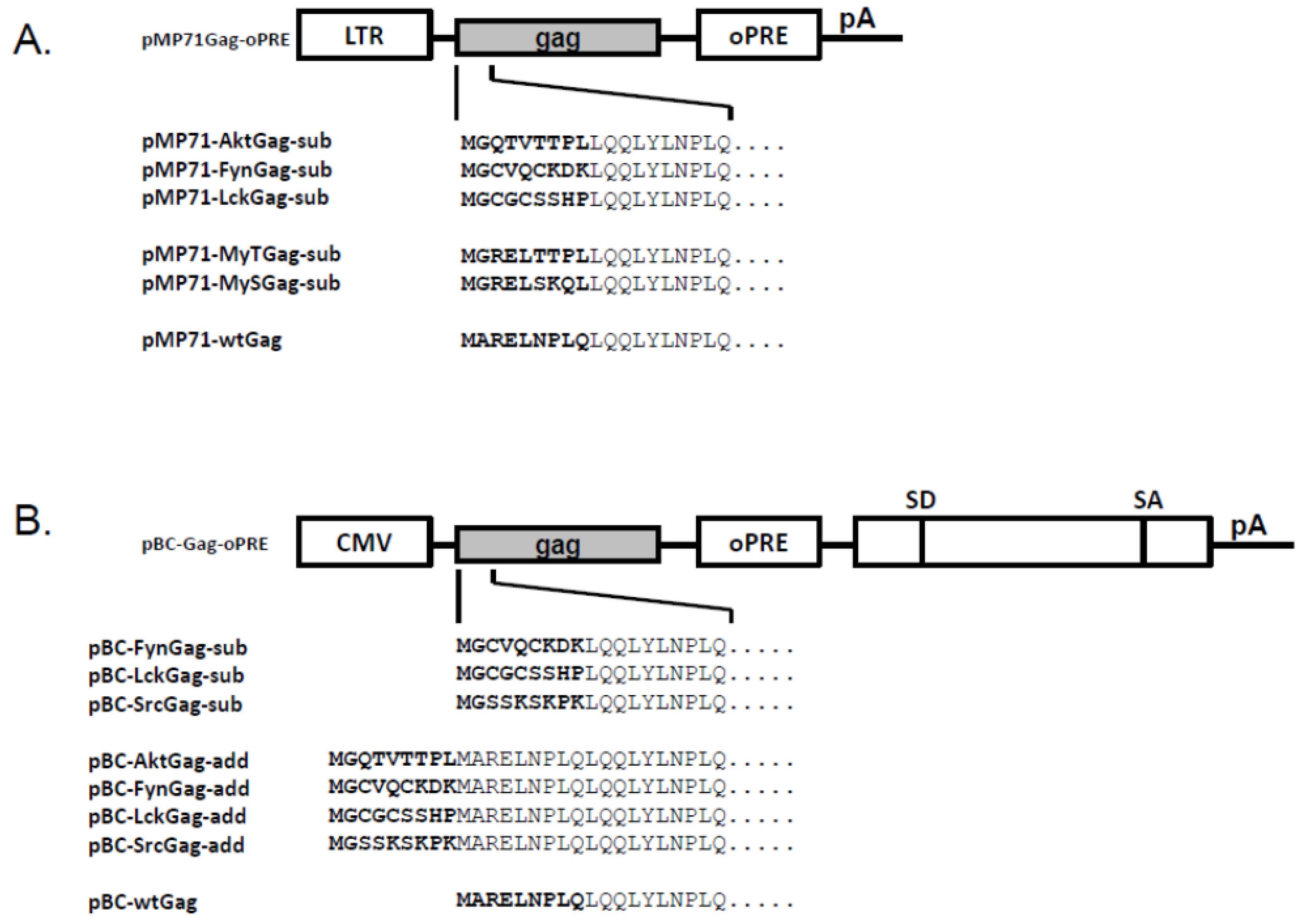
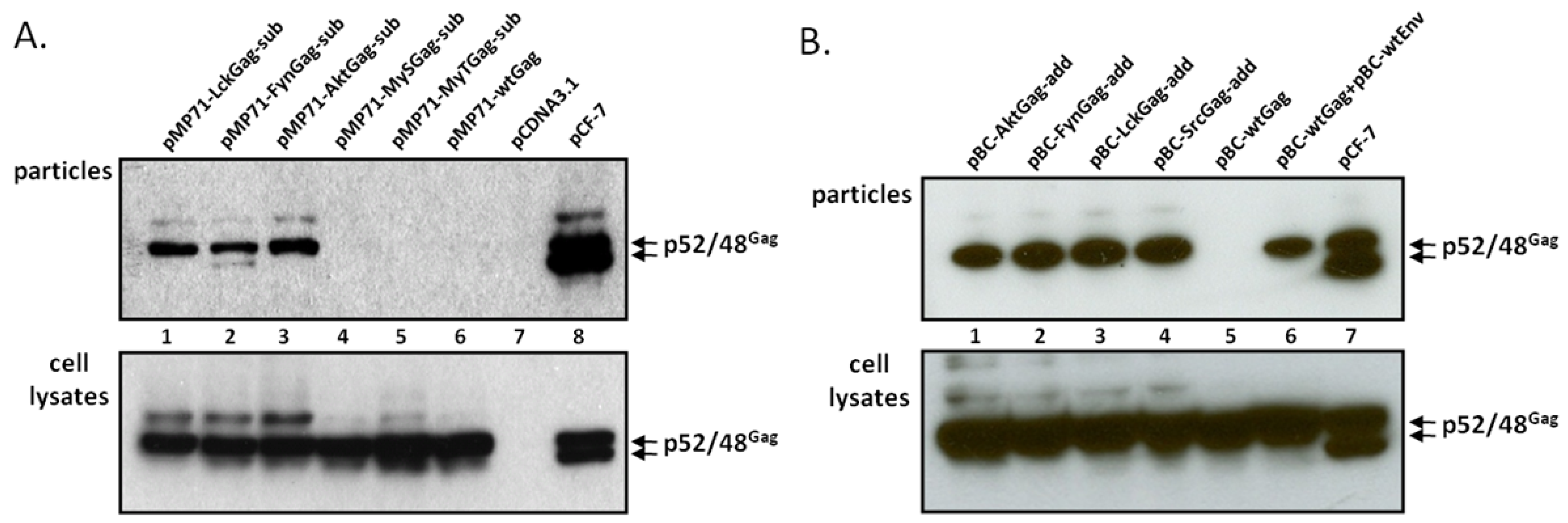
2.1. FFV Gag Myr-Substitution Clones and Particle Budding
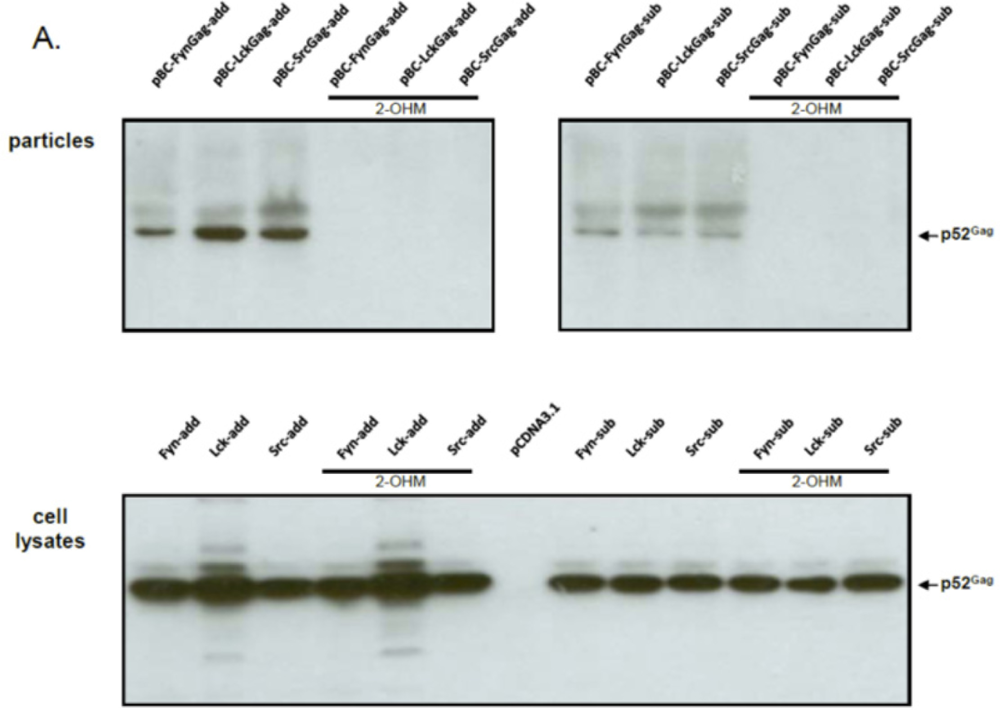
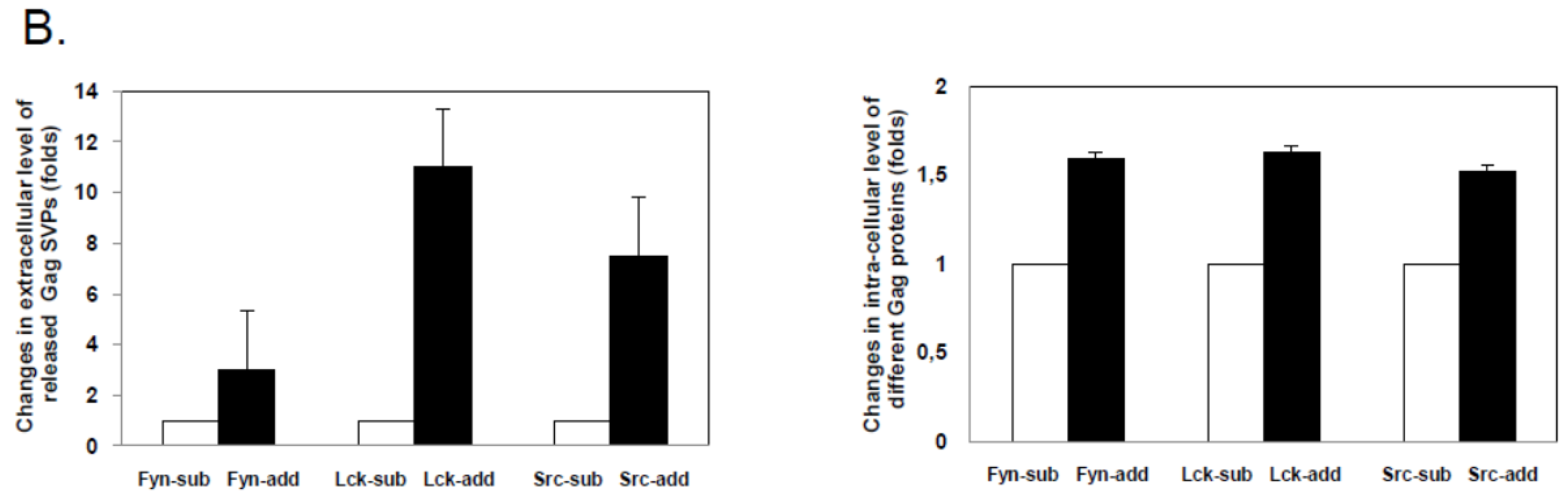
2.2. FFV Gag Myr-Addition Clones and Particle Budding
2.3. The Budding Capacity of FFV Myr-Gag Proteins Is Myristoylation-Dependent
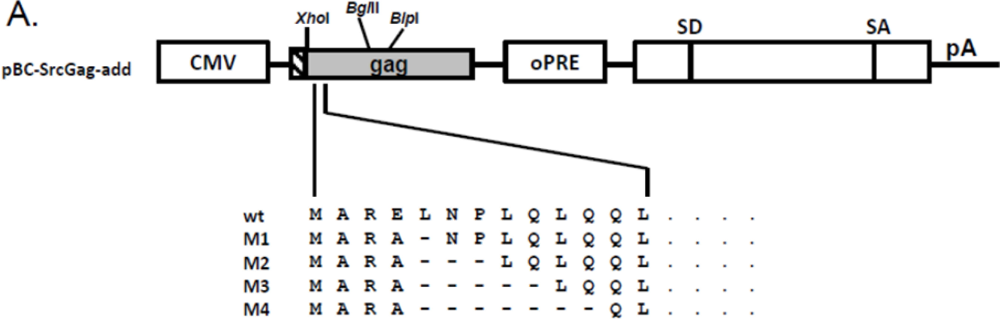
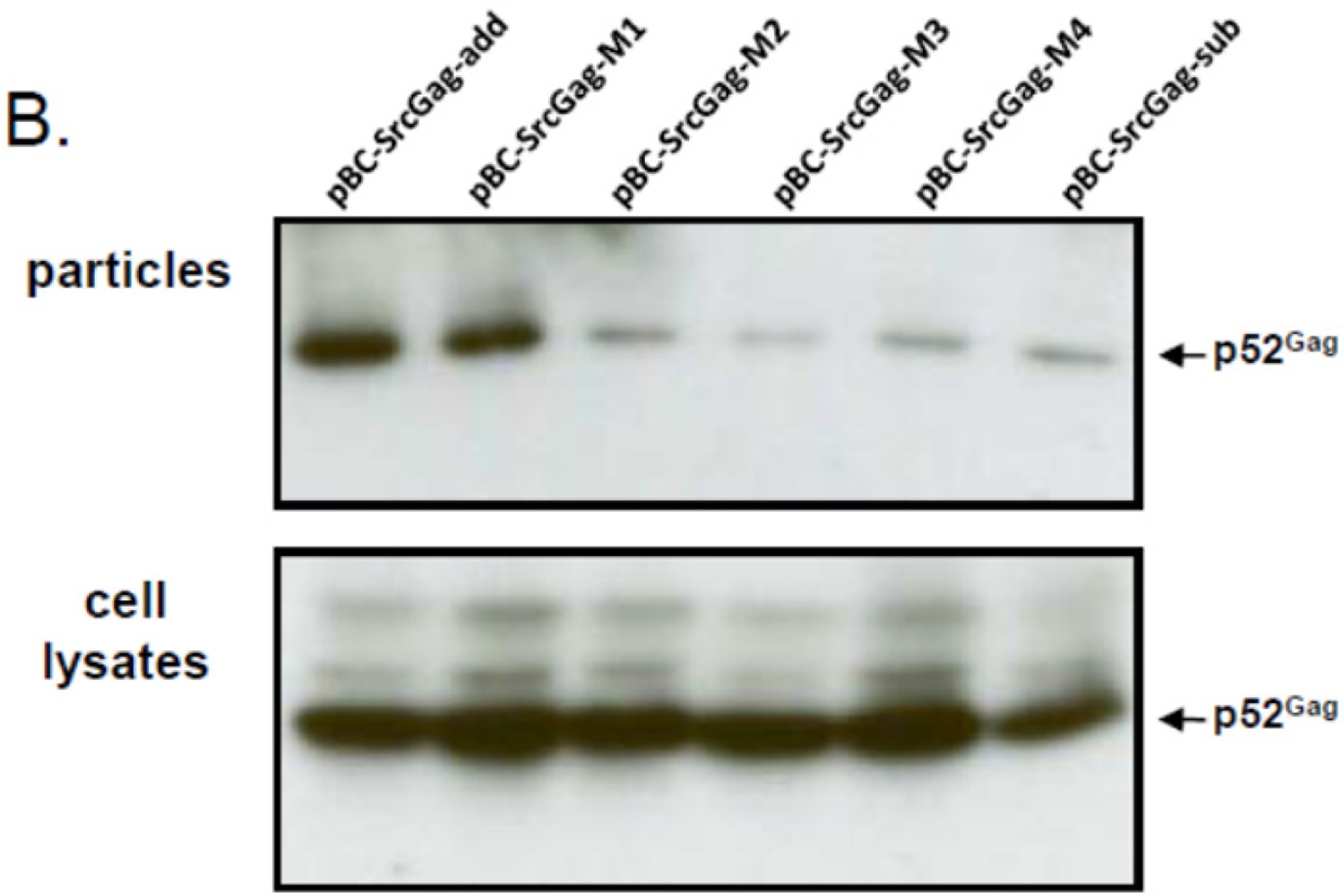
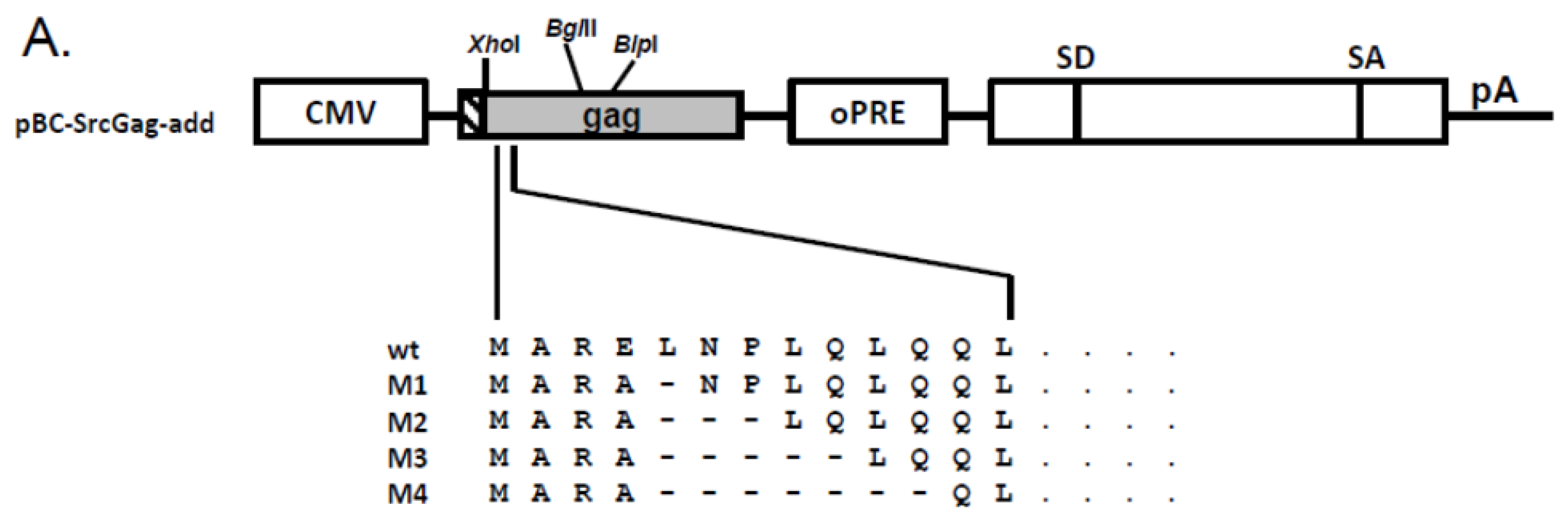
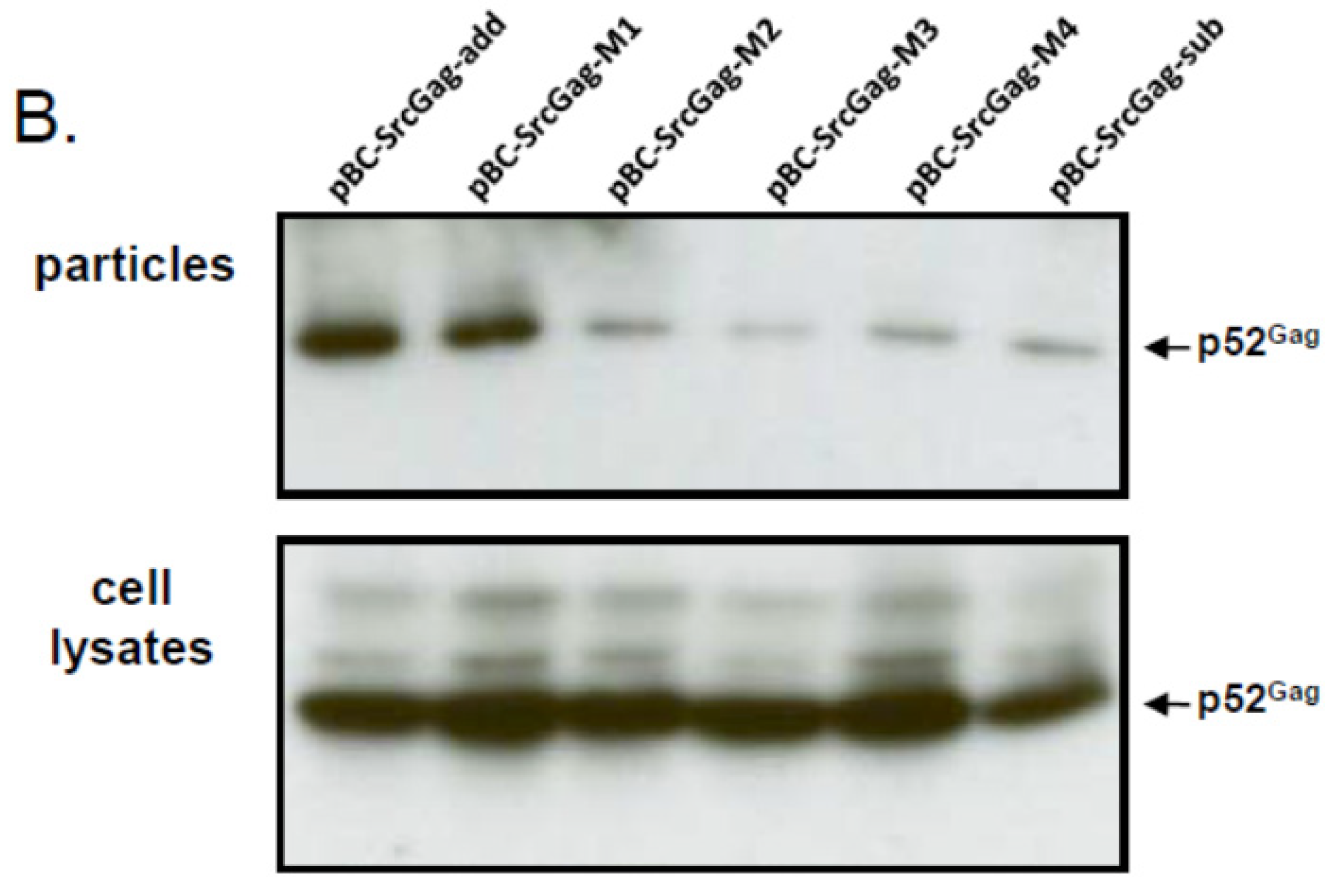
2.5. N-Terminal Deletions in Myr-Gag Proteins Interfere with Budding
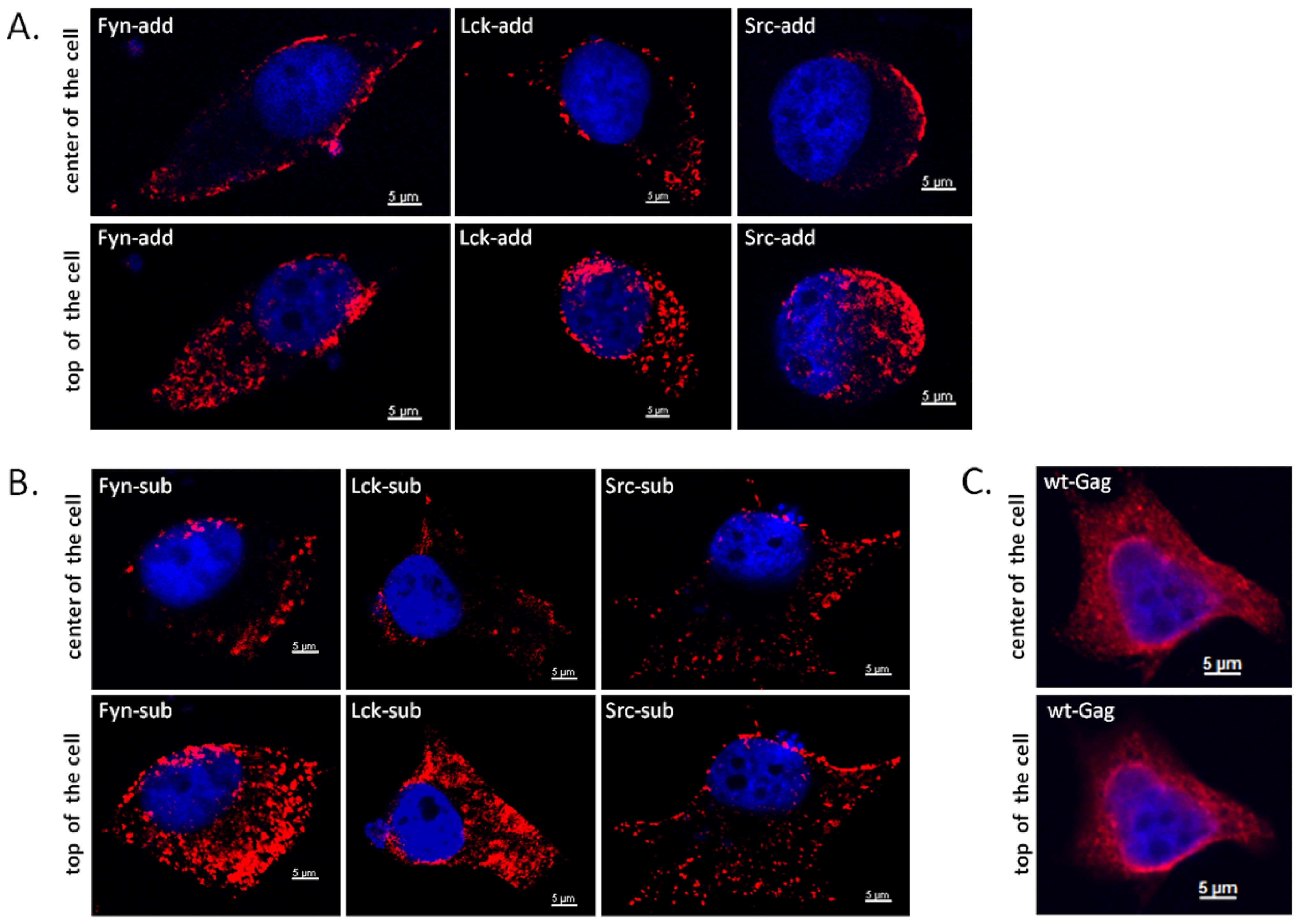
2.6. FFV Myr-Gag Proteins Are Relocalized to the Plasma Membrane in Transfected Cells
3. Experimental Section
3.1. Cell Culture and DNA Transfection
3.2. Purification of FFV Particles and SVPs
3.3. Molecular Cloning of FFV Gag Derivatives
3.4. Protein Detection by Immunoblotting
3.5. IIF and Confocal Laser Microscopy
4. Discussion and Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Assembly, exit, and maturation. In Principles in Virology; American Society for Microbiology: Washington DC, USA, 2009; Volume 1, pp. 453–500. [Google Scholar]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef]
- Dornburg, R. The history and principles of retroviral vectors. Front. Biosci. 2003, 8, d818–835. [Google Scholar] [CrossRef] [PubMed]
- Linial, M. Why aren't foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Linial, M.L. Foamy viruses are unconventional retroviruses. J. Virol. 1999, 73, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Rethwilm, A. Molecular biology of foamy viruses. Med. Microbiol. Immunol. 2010, 199, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef]
- Rethwilm, A. Regulation of foamy virus gene expression. Curr. Top. Microbiol. Immunol. 1995, 193, 1–24. [Google Scholar]
- Rethwilm, A. Foamy virus vectors: An awaited alternative to gammaretro- and lentiviral vectors. Curr. Gene Ther. 2007, 7, 261–271. [Google Scholar] [CrossRef]
- Bastone, P.; Truyen, U.; Löchelt, M. Potential of zoonotic transmission of non-primate foamy viruses to humans. J. Vet. Med. B Infect. Dis. Vet. Public Health 2003, 50, 417–423. [Google Scholar] [CrossRef]
- Heneine, W.; Schweizer, M.; Sandstrom, P.; Folks, T. Human infection with foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 181–196. [Google Scholar]
- Schmidt, M.; Herchenröder, O.; Heeney, J.; Rethwilm, A. Long terminal repeat u3 length polymorphism of human foamy virus. Virology 1997, 230, 167–178. [Google Scholar] [CrossRef]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus env leader protein with the N-terminal gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Bannert, H.; Darai, G.; Flügel, R.M. Analysis of the primary structure of the long terminal repeat and the gag and pol genes of the human spumaretrovirus. J. Virol. 1988, 62, 1590–1597. [Google Scholar] [CrossRef]
- Eastman, S.W.; Linial, M.L. Identification of a conserved residue of foamy virus gag required for intracellular capsid assembly. J. Virol. 2001, 75, 6857–6864. [Google Scholar] [CrossRef]
- Linial, M.L.; Eastman, S.W. Particle assembly and genome packaging. Curr. Top. Microbiol. Immunol. 2003, 277, 89–110. [Google Scholar]
- Yu, S.F.; Eastman, S.W.; Linial, M.L. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in gag. Traffic 2006, 7, 966–977. [Google Scholar] [CrossRef]
- Life, R.B.; Lee, E.G.; Eastman, S.W.; Linial, M.L. Mutations in the amino terminus of foamy virus gag disrupt morphology and infectivity but do not target assembly. J. Virol. 2008, 82, 6109–6119. [Google Scholar] [CrossRef]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 20031–20036. [Google Scholar] [CrossRef]
- Boutin, J.A. Myristoylation. Cell Signal 1997, 9, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Beauchamp, E.; Berthiaume, L.G. Post-translational myristoylation: Fat matters in cellular life and death. Biochimie 2011, 93, 18–31. [Google Scholar] [CrossRef]
- Schambach, A.; Bohne, J.; Baum, C.; Hermann, F.G.; Egerer, L.; von Laer, D.; Giroglou, T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from x protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006, 13, 641–645. [Google Scholar] [CrossRef]
- Liu, W.; Backes, P.; Löchelt, M. Importance of the major splice donor and redefinition of cis-acting sequences of gutless feline foamy virus vectors. Virology 2009, 394, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Doerks, A.; Aboud, M.; Alonso, A.; Tokino, T.; Flügel, R.M.; Löchelt, M. Induction of cellular genes is mediated by the bel1 transactivator in foamy virus-infected human cells. J. Virol. 2000, 74, 4441–4447. [Google Scholar] [CrossRef] [PubMed]
- Wilk, T.; de Haas, F.; Wagner, A.; Rutten, T.; Fuller, S.; Flügel, R.M.; Löchelt, M. The intact retroviral env glycoprotein of human foamy virus is a trimer. J. Virol. 2000, 74, 2885–2887. [Google Scholar] [CrossRef]
- Schwantes, A.; Ortlepp, I.; Löchelt, M. Construction and functional characterization of feline foamy virus-based retroviral vectors. Virology 2002, 301, 53–63. [Google Scholar] [CrossRef]
- Winkler, I.; Bodem, J.; Haas, L.; Zemba, M.; Delius, H.; Flower, R.; Flügel, R.M.; Löchelt, M. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J. Virol. 1997, 71, 6727–6741. [Google Scholar] [CrossRef]
- Alke, A.; Schwantes, A.; Kido, K.; Flötenmeyer, M.; Flügel, R.M.; Löchelt, M. The bet gene of feline foamy virus is required for virus replication. Virology 2001, 287, 310–320. [Google Scholar] [CrossRef]
- ImageJ, version 1.43. NIH: Bethesda, MA, USA, 2010.
- AxioVision, version 4.8.1. Carl Zeiss MicroImaging GmbH: Göttingen, Germany, 2010.
- Kim, Y.-B.; Löchelt, M. Deutsches Krebsforschungszentrum, Heidelberg, Germany. Functional characterization of N-terminal Gag deletions in feline foamy virus. Unpublished work, 2009.
- Geiselhart, V.; Schwantes, A.; Bastone, P.; Frech, M.; Löchelt, M. Features of the env leader protein and the n-terminal gag domain of feline foamy virus important for virus morphogenesis. Virology 2003, 310, 235–244. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence a |
|---|---|
| F-Fyn | 5'-CGAGACCGGTCCGCCATGGGCTGCGTGCAGTGCAAGGAC AAGTTACAGCAACTGTATATA-3' |
| F-Lck | 5'-CGAGACCGGTCCGCCATGGGCTGCGGCTGCAGCAGCCAC CCCTTACAGCAACTGTATATA-3' |
| F-Akt | 5'-CGAGACCGGTCCGCCATGGGCCAGACCGTGACCACCCCC CTGTTACAGCAACTGTATATA-3' |
| F-MyT | 5'-CGAGACCGGTCCGCCATGGGCCGGGAACTGAGCAAGCTG CAATTACAGCAACTGTAT-3' |
| F-MyS | 5'-CGAGACCGGTCCGCCATGGGCTGCGGCTGCAGCAGCCAC CCCTTACAGCAACTGTATATA-3' |
| F-Src | 5'-GCTTACCGGTCCGCCATGGGCAGCAGCAAGAGCAAGCCCAAG TTACAGCAACTGTATATA-3' |
| R-Gag985 | 5'-TGTATAGATTGTTAATGACCCTGG-3' |
| Primer Name | Sequence a |
|---|---|
| 1 | 5'-CCGGTACCGCCATGGGCCAGACCGTGACCACCCCCCTGATGGC-3' |
| 2 | 5'-TCGAGCCATCAGGGGGGTGGTCACGGTCTGGCCCATGGCGGTA-3' |
| 3 | 5'-CCGGTACCGCCATGGGCTGCGTGCAGTGCAAGGACAAGATGGC-3' |
| 4 | 5'-TCGAGCCATCTTGTCCTTGCACTGCACGCAGCCCATGGCGGTA-3' |
| 5 | 5'-CCGGTACCGCCATGGGCTGCGGCTGCAGCAGCCACCCCATGGC-3' |
| 6 | 5'-TCGAGCCATGGGGTGGCTGCTGCAGCCGCAGCCCATGGCGGTA-3' |
| 7 | 5'-CCGGTACCGCCATGGGCAGCAGCAAGAGCAAGCCCAAGATGGC-3' |
| 8 | 5'-TCGAGCCATCTTGGGCTTGCTCTTGCTGCTGCCCATGGCGGTA-3' |
| Construct | Oligonucleotides |
|---|---|
| pBC-AktGag-add | 1,2 |
| pBC-FynGag-add | 3,4 |
| pBC-LckGag-add | 5,6 |
| pBC-SrcGag-add | 7,8 |
| Primer Name | Sequence a |
|---|---|
| M1 | 5'-TGGCTCGAGCGAATCCTCTCCAATTACAG-3' |
| M2 | 5'-TGGCTCGAGCGCTCCAATTACAGCAACTG-3' |
| M3 | 5'-TGGCTCGAGCGTTACAGCAACTGTATATAAA-'3 |
| M4 | 5'-TGGCTCGAGCGCAACTGTATATAAATAATGGC-3' |
| AS-Primer | 5'-CCTAGGTTGAATGCAGTTTGT-3' |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Kim, Y.-B.; Löchelt, M. N-Terminally Myristoylated Feline Foamy Virus Gag Allows Env-Independent Budding of Sub-Viral Particles. Viruses 2011, 3, 2223-2237. https://doi.org/10.3390/v3112223
Liu Y, Kim Y-B, Löchelt M. N-Terminally Myristoylated Feline Foamy Virus Gag Allows Env-Independent Budding of Sub-Viral Particles. Viruses. 2011; 3(11):2223-2237. https://doi.org/10.3390/v3112223
Chicago/Turabian StyleLiu, Yang, Yong-Boum Kim, and Martin Löchelt. 2011. "N-Terminally Myristoylated Feline Foamy Virus Gag Allows Env-Independent Budding of Sub-Viral Particles" Viruses 3, no. 11: 2223-2237. https://doi.org/10.3390/v3112223
APA StyleLiu, Y., Kim, Y. -B., & Löchelt, M. (2011). N-Terminally Myristoylated Feline Foamy Virus Gag Allows Env-Independent Budding of Sub-Viral Particles. Viruses, 3(11), 2223-2237. https://doi.org/10.3390/v3112223