Viral Determinants of HIV-1 Macrophage Tropism

{kind=link}
Abstract
:1. Introduction
2. Beyond Co-Receptor Usage
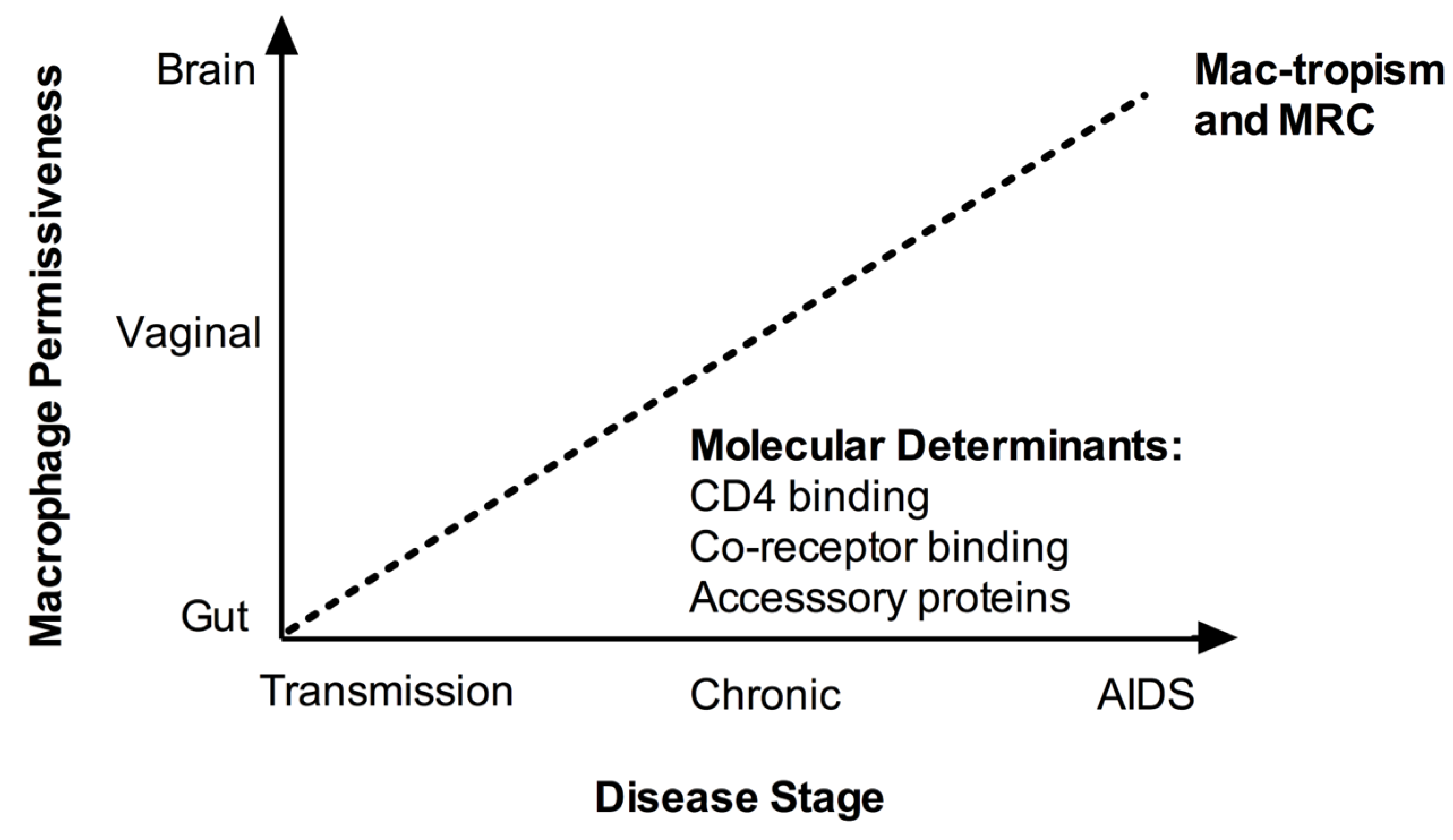
3. Association with Disease Stage
4. Transmitted/Founder Viruses
5. Compartmentalization
6. Influence of Recombination
7. Molecular Determinants — Entry
7.1. CD4 Binding
7.2. Co-Receptor Binding
8. Molecular Determinants — Post Entry
8.1. Tat
8.2. Vif
8.3. Nef
8.4. Vpu
8.5. Vpr/Vpx
9. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Gras, G.; Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 2010, 7, 30. [Google Scholar] [CrossRef]
- Coiras, M.; Lopez-Huertas, M.R.; Perez-Olmeda, M.; Alcami, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Gousset, K.; Ablan, S.D.; Coren, L.V.; Ono, A.; Soheilian, F.; Nagashima, K.; Ott, D.E.; Freed, E.O. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008, 4, e1000015. [Google Scholar] [CrossRef]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef]
- Goodenow, M.M.; Rose, S.L.; Tuttle, D.L.; Sleasman, J.W. HIV-1 fitness and macrophages. J. Leukoc. Biol. 2003, 74, 657–666. [Google Scholar] [CrossRef]
- Gorry, P.R.; Ancuta, P. Coreceptors and HIV-1 pathogenesis. Curr. HIV AIDS Rep. 2011, 8, 45–53. [Google Scholar] [CrossRef]
- Ugolini, S.; Mondor, I.; Sattentau, Q.J. HIV-1 attachment: Another look. Trends Microbiol. 1999, 7, 144–149. [Google Scholar] [CrossRef]
- Ballana, E.; Pauls, E.; Senserrich, J.; Clotet, B.; Perron-Sierra, F.; Tucker, G.C.; Este, J.A. Cell adhesion through alphaV-containing integrins is required for efficient HIV-1 infection in macrophages. Blood 2009, 113, 1278–1286. [Google Scholar] [CrossRef]
- Saphire, A.C.; Bobardt, M.D.; Zhang, Z.; David, G.; Gallay, P.A. Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 2001, 75, 9187–9200. [Google Scholar] [CrossRef]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef]
- Gray, L.; Sterjovski, J.; Churchill, M.; Ellery, P.; Nasr, N.; Lewin, S.R.; Crowe, S.M.; Wesselingh, S.L.; Cunningham, A.L.; Gorry, P.R. Uncoupling coreceptor usage of human immunodeficiency virus type 1 (HIV-1) from macrophage tropism reveals biological properties of CCR5-restricted HIV-1 isolates from patients with acquired immunodeficiency syndrome. Virology 2005, 337, 384–398. [Google Scholar] [CrossRef]
- Peters, P.J.; Sullivan, W.M.; Duenas-Decamp, M.J.; Bhattacharya, J.; Ankghuambom, C.; Brown, R.; Luzuriaga, K.; Bell, J.; Simmonds, P.; Ball, J.; et al. Non-macrophage-tropic human immunodeficiency virus type 1 R5 envelopes predominate in blood, lymph nodes, and semen: Implications for transmission and pathogenesis. J. Virol. 2006, 80, 6324–6332. [Google Scholar] [CrossRef]
- Keele, B.F. Identifying and characterizing recently transmitted viruses. Curr. Opin. HIV AIDS 2010, 5, 327–334. [Google Scholar] [CrossRef]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7552–7557. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, H.; Wilcox, C.K.; van't Wout, A.; Andrus, T.; Llewellyn, N.; Stamatatos, L.; Mullins, J.I.; Corey, L.; Zhu, T. Blood monocytes harbor HIV type 1 strains with diversified phenotypes including macrophage-specific CCR5 virus. J. Infect. Dis. 2008, 197, 309–318. [Google Scholar] [CrossRef]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; Dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar] [CrossRef]
- Smith, P.D.; Fox, C.H.; Masur, H.; Winter, H.S.; Alling, D.W. Quantitative analysis of mononuclear cells expressing human immunodeficiency virus type 1 RNA in esophageal mucosa. J. Exp. Med. 1994, 180, 1541–1546. [Google Scholar] [CrossRef]
- Lewin, S.R.; Kirihara, J.; Sonza, S.; Irving, L.; Mills, J.; Crowe, S.M. HIV-1 DNA and mRNA concentrations are similar in peripheral blood monocytes and alveolar macrophages in HIV-1-infected individuals. AIDS 1998, 12, 719–727. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Pancino, G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology 2010, 7, 31. [Google Scholar] [CrossRef]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef]
- Goodenow, M.M.; Collman, R.G. HIV-1 coreceptor preference is distinct from target cell tropism: A dual-parameter nomenclature to define viral phenotypes. J. Leukoc. Biol. 2006, 80, 965–972. [Google Scholar] [CrossRef]
- Jayakumar, P.; Berger, I.; Autschbach, F.; Weinstein, M.; Funke, B.; Verdin, E.; Goldsmith, M.A.; Keppler, O.T. Tissue-resident macrophages are productively infected ex vivo by primary X4 isolates of human immunodeficiency virus type 1. J. Virol. 2005, 79, 5220–5226. [Google Scholar] [CrossRef]
- Verani, A.; Pesenti, E.; Polo, S.; Tresoldi, E.; Scarlatti, G.; Lusso, P.; Siccardi, A.G.; Vercelli, D. CXCR4 is a functional coreceptor for infection of human macrophages by CXCR4-dependent primary HIV-1 isolates. J. Immunol. 1998, 161, 2084–2088. [Google Scholar] [CrossRef]
- Tuttle, D.L.; Anders, C.B.; Aquino-De Jesus, M.J.; Poole, P.P.; Lamers, S.L.; Briggs, D.R.; Pomeroy, S.M.; Alexander, L.; Peden, K.W.; Andiman, W.A.; et al. Increased replication of non-syncytium-inducing HIV type 1 isolates in monocyte-derived macrophages is linked to advanced disease in infected children. AIDS Res. Hum. Retrovir. 2002, 18, 353–362. [Google Scholar] [CrossRef]
- Li, S.; Juarez, J.; Alali, M.; Dwyer, D.; Collman, R.; Cunningham, A.; Naif, H.M. Persistent CCR5 utilization and enhanced macrophage tropism by primary blood human immunodeficiency virus type 1 isolates from advanced stages of disease and comparison to tissue-derived isolates. J. Virol. 1999, 73, 9741–9755. [Google Scholar] [CrossRef]
- Gorry, P.R.; Sterjovski, J.; Churchill, M.; Witlox, K.; Gray, L.; Cunningham, A.; Wesselingh, S. The role of viral coreceptors and enhanced macrophage tropism in human immunodeficiency virus type 1 disease progression. Sex Health 2004, 1, 23–34. [Google Scholar] [CrossRef]
- Kuroda, M.J. Macrophages: Do they impact AIDS progression more than CD4 T cells? J. Leukoc. Biol. 2010, 87, 569–573. [Google Scholar] [CrossRef]
- Richards, K.H.; Aasa-Chapman, M.M.; McKnight, A.; Clapham, P.R. Modulation of HIV-1 macrophage-tropism among R5 envelopes occurs before detection of neutralizing antibodies. Retrovirology 2010, 7, 48. [Google Scholar] [CrossRef]
- Masciotra, S.; Owen, S.M.; Rudolph, D.; Yang, C.; Wang, B.; Saksena, N.; Spira, T.; Dhawan, S.; Lal, R.B. Temporal relationship between V1V2 variation, macrophage replication, and coreceptor adaptation during HIV-1 disease progression. AIDS 2002, 16, 1887–1898. [Google Scholar] [CrossRef]
- Etemad, B.; Fellows, A.; Kwambana, B.; Kamat, A.; Feng, Y.; Lee, S.; Sagar, M. Human immunodeficiency virus type 1 V1-to-V5 envelope variants from the chronic phase of infection use CCR5 and fuse more efficiently than those from early after infection. J. Virol. 2009, 83, 9694–9708. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Douek, D.C. HIV disease: Fallout from a mucosal catastrophe? Nat. Immunol. 2006, 7, 235–239. [Google Scholar] [CrossRef]
- Hasegawa, A.; Liu, H.; Ling, B.; Borda, J.T.; Alvarez, X.; Sugimoto, C.; Vinet-Oliphant, H.; Kim, W.K.; Williams, K.C.; Ribeiro, R.M.; et al. The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood 2009, 114, 2917–2925. [Google Scholar] [CrossRef]
- Burdo, T.H.; Soulas, C.; Orzechowski, K.; Button, J.; Krishnan, A.; Sugimoto, C.; Alvarez, X.; Kuroda, M.J.; Williams, K.C. Increased monocyte turnover from bone marrow correlates with severity of SIV encephalitis and CD163 levels in plasma. PLoS Pathog. 2010, 6, e1000842. [Google Scholar] [CrossRef]
- Herbein, G.; Varin, A. The macrophage in HIV-1 infection: From activation to deactivation? Retrovirology 2010, 7, 33. [Google Scholar] [CrossRef]
- Wallet, M.A.; Rodriguez, C.A.; Yin, L.; Saporta, S.; Chinratanapisit, S.; Hou, W.; Sleasman, J.W.; Goodenow, M.M. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS 2010, 24, 1281–1290. [Google Scholar] [CrossRef]
- Saez-Cirion, A.; Hamimi, C.; Bergamaschi, A.; David, A.; Versmisse, P.; Melard, A.; Boufassa, F.; Barre-Sinoussi, F.; Lambotte, O.; Rouzioux, C.; et al. Restriction of HIV-1 replication in macrophages and CD4+ T cells from HIV controllers. Blood 2011, 118, 955–964. [Google Scholar] [CrossRef]
- Zhu, T.; Mo, H.; Wang, N.; Nam, D.S.; Cao, Y.; Koup, R.A.; Ho, D.D. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 1993, 261, 1179–1181. [Google Scholar] [CrossRef]
- van't Wout, A.B.; Kootstra, N.A.; Mulder-Kampinga, G.A.; Albrecht-van Lent, N.; Scherpbier, H.J.; Veenstra, J.; Boer, K.; Coutinho, R.A.; Miedema, F.; Schuitemaker, H. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Invest. 1994, 94, 2060–2067. [Google Scholar] [CrossRef]
- Margolis, L.; Shattock, R. Selective transmission of CCR5-utilizing HIV-1: The 'gatekeeper' problem resolved? Nat. Rev. Microbiol. 2006, 4, 312–317. [Google Scholar] [CrossRef]
- Haase, A.T. Targeting early infection to prevent HIV-1 mucosal transmission. Nature 2010, 464, 217–223. [Google Scholar] [CrossRef]
- Nawaz, F.; Cicala, C.; Van Ryk, D.; Block, K.E.; Jelicic, K.; McNally, J.P.; Ogundare, O.; Pascuccio, M.; Patel, N.; Wei, D.; et al. The genotype of early-transmitting HIV gp120s promotes alpha (4) beta(7)-reactivity, revealing alpha (4) beta(7) +/CD4+ T cells as key targets in mucosal transmission. PLoS Pathog. 2011, 7, e1001301. [Google Scholar] [CrossRef]
- Keele, B.F.; Derdeyn, C.A. Genetic and antigenic features of the transmitted virus. Curr. Opin. HIV AIDS 2009, 4, 352–357. [Google Scholar] [CrossRef]
- Abrahams, M.R.; Anderson, J.A.; Giorgi, E.E.; Seoighe, C.; Mlisana, K.; Ping, L.H.; Athreya, G.S.; Treurnicht, F.K.; Keele, B.F.; Wood, N.; et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J. Virol. 2009, 83, 3556–3567. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 2010, 5, e12303. [Google Scholar] [CrossRef]
- Haaland, R.E.; Hawkins, P.A.; Salazar-Gonzalez, J.; Johnson, A.; Tichacek, A.; Karita, E.; Manigart, O.; Mulenga, J.; Keele, B.F.; Shaw, G.M.; et al. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 2009, 5, e1000274. [Google Scholar] [CrossRef]
- Li, H.; Bar, K.J.; Wang, S.; Decker, J.M.; Chen, Y.; Sun, C.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Learn, G.H.; Morgan, C.J.; et al. High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathog. 2010, 6, e1000890. [Google Scholar] [CrossRef]
- Bar, K.J.; Li, H.; Chamberland, A.; Tremblay, C.; Routy, J.P.; Grayson, T.; Sun, C.; Wang, S.; Learn, G.H.; Morgan, C.J.; et al. Wide variation in the multiplicity of HIV-1 infection among injection drug users. J. Virol. 2010, 84, 6241–6247. [Google Scholar] [CrossRef]
- Kishko, M.; Somasundaran, M.; Brewster, F.; Sullivan, J.L.; Clapham, P.R.; Luzuriaga, K. Genotypic and functional properties of early infant HIV-1 envelopes. Retrovirology 2011, 8, 67. [Google Scholar] [CrossRef]
- Isaacman-Beck, J.; Hermann, E.A.; Yi, Y.; Ratcliffe, S.J.; Mulenga, J.; Allen, S.; Hunter, E.; Derdeyn, C.A.; Collman, R.G. Heterosexual transmission of human immunodeficiency virus type 1 subtype C: Macrophage tropism, alternative coreceptor use, and the molecular anatomy of CCR5 utilization. J. Virol. 2009, 83, 8208–8220. [Google Scholar] [CrossRef]
- Ochsenbauer-Jambor, C.; Ding, H.; Keele, B.F.; Salazar-Gonzalez, J.; Edmonds, T.G.; Shattock, R.; Clapham, P.; Shaw, G.; Hahn, B.H.; Kappes, J.C. Generation and biological characterization of infectious molecular clones derived from clade b HIV-1 transmitted/founder viruses. In Proceedings of the 16th Conference on Retroviruses and Opportunistic Infections, Montreal, Canada, 8–11 February 2009. Session 89, No. 492. [Google Scholar]
- Baalwa, J.; Wang, S.; Yue, L.; Kaleebu, P.; Cormier, E.; Gilmour, J.; Hunter, E.; Haynes, B.F.; Hahn, B.H.; Shaw, G. Enhanced fusogenecity, replication kinetics, and macrophage tropism of subtype d transmitted/founder viruses in acute HIV-1 infection. In Proceedings of the 18th Conference on Retroviruses and Opportunistic Infections, Boston, MA, 27 February–2 March 2011. Session 102, No. 495. [Google Scholar]
- Guo, J.; Wang, W.; Yu, D.; Wu, Y. Spinoculation triggers dynamic actin and cofilin activity facilitating HIV-1 infection of transformed and resting CD4 T cells. J. Virol. 2011, 85, 9824–9833. [Google Scholar] [CrossRef]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef]
- Ghorpade, A.; Nukuna, A.; Che, M.; Haggerty, S.; Persidsky, Y.; Carter, E.; Carhart, L.; Shafer, L.; Gendelman, H.E. Human immunodeficiency virus neurotropism: An analysis of viral replication and cytopathicity for divergent strains in monocytes and microglia. J. Virol. 1998, 72, 3340–3350. [Google Scholar] [CrossRef]
- Gorry, P.R.; Bristol, G.; Zack, J.A.; Ritola, K.; Swanstrom, R.; Birch, C.J.; Bell, J.E.; Bannert, N.; Crawford, K.; Wang, H.; et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J. Virol. 2001, 75, 10073–10089. [Google Scholar] [CrossRef]
- Smit, T.K.; Wang, B.; Ng, T.; Osborne, R.; Brew, B.; Saksena, N.K. Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): Evidence for neurotropic HIV variants. Virology 2001, 279, 509–526. [Google Scholar] [CrossRef]
- Peters, P.J.; Bhattacharya, J.; Hibbitts, S.; Dittmar, M.T.; Simmons, G.; Bell, J.; Simmonds, P.; Clapham, P.R. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J. Virol. 2004, 78, 6915–6926. [Google Scholar]
- Dunfee, R.L.; Thomas, E.R.; Gorry, P.R.; Wang, J.; Taylor, J.; Kunstman, K.; Wolinsky, S.M.; Gabuzda, D. The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15160–15165. [Google Scholar] [CrossRef]
- Dunfee, R.L.; Thomas, E.R.; Wang, J.; Kunstman, K.; Wolinsky, S.M.; Gabuzda, D. Loss of the n-linked glycosylation site at position 386 in the HIV envelope V4 region enhances macrophage tropism and is associated with dementia. Virology 2007, 367, 222–234. [Google Scholar] [CrossRef]
- Peters, P.J.; Duenas-Decamp, M.J.; Sullivan, W.M.; Clapham, P.R. Variation of macrophage tropism among HIV-1 R5 envelopes in brain and other tissues. J. Neuroimmune Pharmacol. 2007, 2, 32–41. [Google Scholar] [CrossRef]
- Thomas, E.R.; Dunfee, R.L.; Stanton, J.; Bogdan, D.; Taylor, J.; Kunstman, K.; Bell, J.E.; Wolinsky, S.M.; Gabuzda, D. Macrophage entry mediated by HIV Envs from brain and lymphoid tissues is determined by the capacity to use low CD4 levels and overall efficiency of fusion. Virology 2007, 360, 105–119. [Google Scholar] [CrossRef]
- Rossi, F.; Querido, B.; Nimmagadda, M.; Cocklin, S.; Navas-Martin, S.; Martin-Garcia, J. The V1-V3 region of a brain-derived HIV-1 envelope glycoprotein determines macrophage tropism, low CD4 dependence, increased fusogenicity and altered sensitivity to entry inhibitors. Retrovirology 2008, 5, 89. [Google Scholar] [CrossRef]
- Dunfee, R.L.; Thomas, E.R.; Gabuzda, D. Enhanced macrophage tropism of HIV in brain and lymphoid tissues is associated with sensitivity to the broadly neutralizing CD4 binding site antibody b12. Retrovirology 2009, 6, 69. [Google Scholar] [CrossRef]
- Gray, L.; Roche, M.; Churchill, M.J.; Sterjovski, J.; Ellett, A.; Poumbourios, P.; Sherieff, S.; Wang, B.; Saksena, N.; Purcell, D.F.; et al. Tissue-specific sequence alterations in the human immunodeficiency virus type 1 envelope favoring CCR5 usage contribute to persistence of dual-tropic virus in the brain. J. Virol. 2009, 83, 5430–5441. [Google Scholar] [CrossRef]
- van Marle, G.; Gill, M.J.; Kolodka, D.; McManus, L.; Grant, T.; Church, D.L. Compartmentalization of the gut viral reservoir in HIV-1 infected patients. Retrovirology 2007, 4, 87. [Google Scholar] [CrossRef]
- Tovanabutra, S.; de Souza, M.; Sittisombut, N.; Sriplienchan, S.; Ketsararat, V.; Birx, D.L.; Khamboonrueng, C.; Nelson, K.E.; McCutchan, F.E.; Robb, M.L. HIV-1 genetic diversity and compartmentalization in mother/infant pairs infected with CRF01_AE. AIDS 2007, 21, 1050–1053. [Google Scholar] [CrossRef]
- Ritola, K.; Robertson, K.; Fiscus, S.A.; Hall, C.; Swanstrom, R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J. Virol. 2005, 79, 10830–10834. [Google Scholar] [CrossRef]
- Burkala, E.J.; He, J.; West, J.T.; Wood, C.; Petito, C.K. Compartmentalization of HIV-1 in the central nervous system: Role of the choroid plexus. AIDS 2005, 19, 675–684. [Google Scholar] [CrossRef]
- Abbate, I.; Cappiello, G.; Longo, R.; Ursitti, A.; Spano, A.; Calcaterra, S.; Dianzani, F.; Antinori, A.; Capobianchi, M.R. Cell membrane proteins and quasispecies compartmentalization of CSF and plasma HIV-1 from AIDS patients with neurological disorders. Infect. Genet. Evol. 2005, 5, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kemal, K.S.; Foley, B.; Burger, H.; Anastos, K.; Minkoff, H.; Kitchen, C.; Philpott, S.M.; Gao, W.; Robison, E.; Holman, S.; et al. HIV-1 in genital tract and plasma of women: Compartmentalization of viral sequences, coreceptor usage, and glycosylation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12972–12977. [Google Scholar] [CrossRef] [PubMed]
- Becquart, P.; Chomont, N.; Roques, P.; Ayouba, A.; Kazatchkine, M.D.; Belec, L.; Hocini, H. Compartmentalization of HIV-1 between breast milk and blood of HIV-infected mothers. Virology 2002, 300, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Marras, D.; Bruggeman, L.A.; Gao, F.; Tanji, N.; Mansukhani, M.M.; Cara, A.; Ross, M.D.; Gusella, G.L.; Benson, G.; D'Agati, V.D.; et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat. Med. 2002, 8, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.N.; Birch, C.; Middleton, T.; Dwyer, D.E.; Cunningham, A.L.; Saksena, N.K. Evidence for late stage compartmentalization of HIV-1 resistance mutations between lymph node and peripheral blood mononuclear cells. AIDS 2000, 14, 2273–2281. [Google Scholar] [CrossRef]
- Coombs, R.W.; Speck, C.E.; Hughes, J.P.; Lee, W.; Sampoleo, R.; Ross, S.O.; Dragavon, J.; Peterson, G.; Hooton, T.M.; Collier, A.C.; et al. Association between culturable human immunodeficiency virus type 1 (HIV-1) in semen and HIV-1 RNA levels in semen and blood: Evidence for compartmentalization of HIV-1 between semen and blood. J. Infect. Dis. 1998, 177, 320–330. [Google Scholar] [CrossRef]
- Fulcher, J.A.; Hwangbo, Y.; Zioni, R.; Nickle, D.; Lin, X.; Heath, L.; Mullins, J.I.; Corey, L.; Zhu, T. Compartmentalization of human immunodeficiency virus type 1 between blood monocytes and CD4+ T cells during infection. J. Virol. 2004, 78, 7883–7893. [Google Scholar] [CrossRef]
- Potter, S.J.; Lemey, P.; Achaz, G.; Chew, C.B.; Vandamme, A.M.; Dwyer, D.E.; Saksena, N.K. HIV-1 compartmentalization in diverse leukocyte populations during antiretroviral therapy. J. Leukoc. Biol. 2004, 76, 562–570. [Google Scholar] [CrossRef]
- Llewellyn, N.; Zioni, R.; Zhu, H.; Andrus, T.; Xu, Y.; Corey, L.; Zhu, T. Continued evolution of HIV-1 circulating in blood monocytes with antiretroviral therapy: Genetic analysis of HIV-1 in monocytes and CD4+ T cells of patients with discontinued therapy. J. Leukoc. Biol. 2006, 80, 1118–1126. [Google Scholar] [CrossRef]
- Bull, M.; Learn, G.; Genowati, I.; McKernan, J.; Hitti, J.; Lockhart, D.; Tapia, K.; Holte, S.; Dragavon, J.; Coombs, R.; et al. Compartmentalization of HIV-1 within the female genital tract is due to monotypic and low-diversity variants not distinct viral populations. PLoS One 2009, 4, e7122. [Google Scholar] [CrossRef]
- Bull, M.E.; Learn, G.H.; McElhone, S.; Hitti, J.; Lockhart, D.; Holte, S.; Dragavon, J.; Coombs, R.W.; Mullins, J.I.; Frenkel, L.M. Monotypic human immunodeficiency virus type 1 genotypes across the uterine cervix and in blood suggest proliferation of cells with provirus. J. Virol. 2009, 83, 6020–6028. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Ping, L.H.; Dibben, O.; Jabara, C.B.; Arney, L.; Kincer, L.; Tang, Y.; Hobbs, M.; Hoffman, I.; Kazembe, P.; et al. HIV-1 populations in semen arise through multiple mechanisms. PLoS Pathog. 2010, 6, e1001053. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Carlsson, J.; Heath, L.; Bull, M.E.; Shetty, A.K.; Mutsvangwa, J.; Musingwini, G.; Woelk, G.; Zijenah, L.S.; Katzenstein, D.A.; et al. Genetic analyses of HIV-1 env sequences demonstrate limited compartmentalization in breast milk and suggest viral replication within the breast that increases with mastitis. J. Virol. 2010, 84, 10812–10819. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Learn, G.H.; Fouda, G.G.; Kang, H.H.; Mahlokozera, T.; Wilks, A.B.; Lovingood, R.V.; Stacey, A.; Kalilani, L.; et al. Origin and evolution of HIV-1 in breast milk determined by single-genome amplification and sequencing. J. Virol. 2011, 85, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.E.; Poli, G.; Schnittman, S.M.; Psallidopoulos, M.C.; Davey, V.; Fauci, A.S. In vivo T lymphocyte origin of macrophage-tropic strains of HIV. Role of monocytes during in vitro isolation and in vivo infection. J. Immunol. 1990, 144, 4628–4632. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, H.; Wilcox, C.K.; van't Wout, A.; Andrus, T.; Llewellyn, N.; Stamatatos, L.; Mullins, J.I.; Corey, L.; Zhu, T. Blood monocytes harbor HIV type 1 strains with diversified phenotypes including macrophage-specific CCR5 virus. J. Infect. Dis. 2008, 197, 309–318. [Google Scholar] [CrossRef]
- Harrington, P.R.; Schnell, G.; Letendre, S.L.; Ritola, K.; Robertson, K.; Hall, C.; Burch, C.L.; Jabara, C.B.; Moore, D.T.; Ellis, R.J.; et al. Cross-sectional characterization of HIV-1 env compartmentalization in cerebrospinal fluid over the full disease course. AIDS 2009, 23, 907–915. [Google Scholar] [CrossRef]
- Schnell, G.; Price, R.W.; Swanstrom, R.; Spudich, S. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J. Virol. 2010, 84, 2395–2407. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.; Zarate, S.; Smith, D.M.; Little, S.J.; Pillai, S.K.; Ellis, R.J.; Wong, J.K.; Leigh Brown, A.J.; Richman, D.D.; et al. Estimating selection pressures on HIV-1 using phylogenetic likelihood models. Stat. Med. 2008, 27, 4779–4789. [Google Scholar] [CrossRef]
- Collini, P.; Noursadeghi, M.; Sabroe, I.; Miller, R.F.; Dockrell, D.H. Monocyte and macrophage dysfunction as a cause of HIV-1 induced dysfunction of innate immunity. Curr. Mol. Med. 2010, 10, 727–740. [Google Scholar] [CrossRef]
- Shen, R.; Richter, H.E.; Clements, R.H.; Novak, L.; Huff, K.; Bimczok, D.; Sankaran-Walters, S.; Dandekar, S.; Clapham, P.R.; Smythies, L.E.; et al. Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J. Virol. 2009, 83, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Meng, G.; Ochsenbauer, C.; Clapham, P.R.; Grams, J.; Novak, L.; Kappes, J.C.; Smythies, L.E.; Smith, P.D. Stromal down-regulation of macrophage CD4/CCR5 expression and NF-kappaB activation mediates HIV-1 non-permissiveness in intestinal macrophages. PLoS Pathog. 2011, 7, e1002060. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Aasa-Chapman, M.M.; Seymour, C.R.; Williams, I.; McKnight, A. Novel envelope determinants for CCR3 use by human immunodeficiency virus. J. Virol. 2006, 80, 10884–10889. [Google Scholar] [CrossRef]
- Borderia, A.V.; Codoner, F.M.; Sanjuan, R. Selection promotes organ compartmentalization in HIV-1: Evidence from gag and pol genes. Evolution 2007, 61, 272–279. [Google Scholar] [CrossRef]
- Wodarz, D.; Levy, D.N. Effect of different modes of viral spread on the dynamics of multiply infected cells in human immunodeficiency virus infection. J. R. Soc. Interface 2011, 8, 289–300. [Google Scholar] [CrossRef]
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4204–4209. [Google Scholar] [CrossRef]
- Chen, J.; Rhodes, T.D.; Hu, W.S. Comparison of the genetic recombination rates of human immunodeficiency virus type 1 in macrophages and T cells. J. Virol. 2005, 79, 9337–9340. [Google Scholar] [CrossRef]
- Brown, R.J.; Peters, P.J.; Caron, C.; Gonzalez-Perez, M.P.; Stones, L.; Ankghuambom, C.; Pondei, K.; McClure, C.P.; Alemnji, G.; Taylor, S.; et al. Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J. Virol. 2011, 85, 6024–6037. [Google Scholar] [CrossRef]
- Sigal, A.; Kim, J.T.; Balazs, A.B.; Dekel, E.; Mayo, A.; Milo, R.; Baltimore, D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011, 477, 95–98. [Google Scholar] [CrossRef]
- Del Portillo, A.; Tripodi, J.; Najfeld, V.; Wodarz, D.; Levy, D.N.; Chen, B.K. Multiploid inheritance of HIV-1 during cell-to-cell infection. J. Virol. 2011, 85, 7169–7176. [Google Scholar] [CrossRef] [PubMed]
- Lamers, S.L.; Gray, R.R.; Salemi, M.; Huysentruyt, L.C.; McGrath, M.S. HIV-1 phylogenetic analysis shows HIV-1 transits through the meninges to brain and peripheral tissues. Infect. Genet. Evol. 2011, 11, 31–37. [Google Scholar] [CrossRef]
- Peters, P.J.; Duenas-Decamp, M.J.; Sullivan, W.M.; Brown, R.; Ankghuambom, C.; Luzuriaga, K.; Robinson, J.; Burton, D.R.; Bell, J.; Simmonds, P.; et al. Variation in HIV-1 R5 macrophage-tropism correlates with sensitivity to reagents that block envelope: CD4 interactions but not with sensitivity to other entry inhibitors. Retrovirology 2008, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Decamp, M.J.; Peters, P.J.; Burton, D.; Clapham, P.R. Determinants flanking the CD4 binding loop modulate macrophage tropism of human immunodeficiency virus type 1 R5 envelopes. J. Virol. 2009, 83, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Dunfee, R.L.; Thomas, E.R.; Gorry, P.R.; Wang, J.; Taylor, J.; Kunstman, K.; Wolinsky, S.M.; Gabuzda, D. The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15160–15165. [Google Scholar] [CrossRef]
- Duenas-Decamp, M.J.; Peters, P.; Burton, D.; Clapham, P.R. Natural resistance of human immunodeficiency virus type 1 to the CD4BS antibody B12 conferred by a glycan and an arginine residue close to the CD4 binding loop. J. Virol. 2008, 82, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Musich, T.; Peters, P.J.; Duenas-Decamp, M.J.; Gonzalez-Perez, M.P.; Robinson, J.; Zolla-Pazner, S.; Ball, J.K.; Luzuriaga, K.; Clapham, P.R. A conserved determinant in the V1 loop of HIV-1 that modulates the V3 loop to prime low CD4 use and macrophage infection. J. Virol. 2011, 85, 2397–2405. [Google Scholar] [CrossRef]
- Sterjovski, J.; Roche, M.; Churchill, M.J.; Ellett, A.; Farrugia, W.; Gray, L.R.; Cowley, D.; Poumbourios, P.; Lee, B.; Wesselingh, S.L.; et al. An altered and more efficient mechanism of CCR5 engagement contributes to macrophage tropism of CCR5-using HIV-1 envelopes. Virology 2010, 404, 269–278. [Google Scholar] [CrossRef]
- Gray, L.; Roche, M.; Churchill, M.J.; Sterjovski, J.; Ellett, A.; Poumbourios, P.; Sherieff, S.; Wang, B.; Saksena, N.; Purcell, D.F.; et al. Tissue-specific sequence alterations in the human immunodeficiency virus type 1 envelope favoring CCR5 usage contribute to persistence of dual-tropic virus in the brain. J. Virol. 2009, 83, 5430–5441. [Google Scholar] [CrossRef]
- Cashin, K.; Roche, M.; Sterjovski, J.; Ellett, A.; Gray, L.R.; Cunningham, A.L.; Ramsland, P.A.; Churchill, M.J.; Gorry, P.R. Alternative coreceptor requirements for efficient CCR5- and CXCR4-mediated HIV-1 entry into macrophages. J. Virol. 2011, 85, 10699–10709. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14572–14579. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Dayton, A.I.; Sodroski, J.G.; Rosen, C.A.; Goh, W.C.; Haseltine, W.A. The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell 1986, 44, 941–947. [Google Scholar] [CrossRef]
- Sonza, S.; Mutimer, H.P.; O'Brien, K.; Ellery, P.; Howard, J.L.; Axelrod, J.H.; Deacon, N.J.; Crowe, S.M.; Purcell, D.F. Selectively reduced tat mRNA heralds the decline in productive human immunodeficiency virus type 1 infection in monocyte-derived macrophages. J. Virol. 2002, 76, 12611–12621. [Google Scholar] [CrossRef]
- Dowling, D.; Nasr-Esfahani, S.; Tan, C.H.; O'Brien, K.; Howard, J.L.; Jans, D.A.; Purcell, D.F.; Stoltzfus, C.M.; Sonza, S. HIV-1 infection induces changes in expression of cellular splicing factors that regulate alternative viral splicing and virus production in macrophages. Retrovirology 2008, 5, 18. [Google Scholar] [CrossRef]
- Neuveut, C.; Scoggins, R.M.; Camerini, D.; Markham, R.B.; Jeang, K.T. Requirement for the second coding exon of Tat in the optimal replication of macrophage-tropic HIV-1. J. Biomed. Sci. 2003, 10, 651–660. [Google Scholar] [CrossRef]
- Hiebenthal-Millow, K.; Greenough, T.C.; Bretttler, D.B.; Schindler, M.; Wildum, S.; Sullivan, J.L.; Kirchhoff, F. Alterations in HIV-1 LTR promoter activity during AIDS progression. Virology 2003, 317, 109–118. [Google Scholar] [CrossRef]
- Cowley, D.; Gray, L.R.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. Genetic and functional heterogeneity of CNS-derived tat alleles from patients with HIV-associated dementia. J. Neurovirol. 2011, 17, 70–81. [Google Scholar] [CrossRef]
- Zhang, M.; Drenkow, J.; Lankford, C.S.; Frucht, D.M.; Rabin, R.L.; Gingeras, T.R.; Venkateshan, C.; Schwartzkopff, F.; Clouse, K.A.; Dayton, A.I. HIV regulation of the IL-7R: A viral mechanism for enhancing HIV-1 replication in human macrophages in vitro. J. Leukoc. Biol. 2006, 79, 1328–1338. [Google Scholar] [CrossRef]
- Gabuzda, D.H.; Li, H.; Lawrence, K.; Vasir, B.S.; Crawford, K.; Langhoff, E. Essential role of vif in establishing productive HIV-1 infection in peripheral blood T lymphocytes and monocyte/macrophages. J. Acquir. Immune Defic. Syndr. 1994, 7, 908–915. [Google Scholar] [PubMed]
- Kawamura, M.; Ishizaki, T.; Ishimoto, A.; Shioda, T.; Kitamura, T.; Adachi, A. Growth ability of human immunodeficiency virus type 1 auxiliary gene mutants in primary blood macrophage cultures. J. Gen. Virol. 1994, 75, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.H.; Chao, W.; Potash, M.J.; Sova, P.; Gendelman, H.E.; Volsky, D.J. Vif-negative human immunodeficiency virus type 1 persistently replicates in primary macrophages, producing attenuated progeny virus. J. Virol. 1996, 70, 5336–5345. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 675–687. [Google Scholar] [CrossRef]
- Miyagi, E.; Schwartzkopff, F.; Plishka, R.; Buckler-White, A.; Clouse, K.A.; Strebel, K. APOBEC3G-independent reduction in virion infectivity during long-term HIV-1 replication in terminally differentiated macrophages. Virology 2008, 379, 266–274. [Google Scholar] [CrossRef]
- Deacon, N.J.; Tsykin, A.; Solomon, A.; Smith, K.; Ludford-Menting, M.; Hooker, D.J.; McPhee, D.A.; Greenway, A.L.; Ellett, A.; Chatfield, C.; et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 1995, 270, 988–991. [Google Scholar] [CrossRef]
- Kestler, H.W., 3rd; Ringler, D.J.; Mori, K.; Panicali, D.L.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991, 65, 651–662. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Brief report: Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. New Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef]
- Malykh, A.; Reitz, M.S., Jr.; Louie, A.; Hall, L.; Lori, F. Multiple determinants for growth of human immunodeficiency virus type 1 in monocyte-macrophages. Virology 1995, 206, 646–650. [Google Scholar] [CrossRef]
- Miller, M.D.; Warmerdam, M.T.; Gaston, I.; Greene, W.C.; Feinberg, M.B. The human immunodeficiency virus-1 nef gene product: A positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 1994, 179, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Learmont, J.; Tindall, B.; Evans, L.; Cunningham, A.; Cunningham, P.; Wells, J.; Penny, R.; Kaldor, J.; Cooper, D.A. Long-term symptomless HIV-1 infection in recipients of blood products from a single donor. Lancet 1992, 340, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.R.; McPhee, D.A.; Wesselingh, S.L.; Churchill, M.J. Macrophage tropism and cytopathicity of HIV-1 variants isolated sequentially from a long-term survivor infected with nef-deleted virus. Open Microbiol. J. 2007, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Moghaddam, S.; Kawano, T.; Cheng-Mayer, C. Multiple human immunodeficiency virus type 1 Nef functions contribute to efficient replication in primary human macrophages. J. Gen. Virol. 2004, 85, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.A.; Warmerdam, M.T.; Atchison, R.E.; Miller, M.D.; Greene, W.C. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 nef. J. Virol. 1995, 69, 4112–4121. [Google Scholar] [CrossRef]
- Lundquist, C.A.; Tobiume, M.; Zhou, J.; Unutmaz, D.; Aiken, C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J. Virol. 2002, 76, 4625–4633. [Google Scholar] [CrossRef]
- Glushakova, S.; Munch, J.; Carl, S.; Greenough, T.C.; Sullivan, J.L.; Margolis, L.; Kirchhoff, F. CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4(+) T-cell depletion in human lymphoid tissue ex vivo. J. Virol. 2001, 75, 10113–10117. [Google Scholar] [CrossRef]
- Cortes, M.J.; Wong-Staal, F.; Lama, J. Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cells. J. Biol. Chem. 2002, 277, 1770–1779. [Google Scholar] [CrossRef]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. 1999, 9, 613–621. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Bennett, A.E.; Narayan, K.; Shi, D.; Hartnell, L.M.; Gousset, K.; He, H.; Lowekamp, B.C.; Yoo, T.S.; Bliss, D.; Freed, E.O.; et al. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 2009, 5, e1000591. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Groot, F.; Krausslich, H.G.; Keppler, O.T.; Sattentau, Q.J. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J. Virol. 2011, 85, 7922–7927. [Google Scholar] [CrossRef]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Krausslich, H.G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, e36. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.R.; Gabuzda, D.; Cowley, D.; Ellett, A.; Chiavaroli, L.; Wesselingh, S.L.; Churchill, M.J.; Gorry, P.R. CD4 and MHC class 1 down-modulation activities of nef alleles from brain- and lymphoid tissue-derived primary HIV-1 isolates. J. Neurovirol. 2011, 17, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Pushker, R.; Jacque, J.M.; Shields, D.C. Meta-analysis to test the association of HIV-1 nef amino acid differences and deletions with disease progression. J. Virol. 2010, 84, 3644–3653. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Schubert, U.; Clouse, K.A.; Strebel, K. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J. Virol. 1995, 69, 7699–7711. [Google Scholar] [CrossRef]
- Schindler, M.; Rajan, D.; Banning, C.; Wimmer, P.; Koppensteiner, H.; Iwanski, A.; Specht, A.; Sauter, D.; Dobner, T.; Kirchhoff, F. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology 2010, 7, 1. [Google Scholar] [CrossRef]
- Schubert, U.; Bour, S.; Willey, R.L.; Strebel, K. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either vpu or env. J. Virol. 1999, 73, 887–896. [Google Scholar] [CrossRef]
- Richards, K.H.; Clapham, P.R. Effects of vpu start-codon mutations on human immunodeficiency virus type 1 replication in macrophages. J. Gen. Virol. 2007, 88, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Ayinde, D.; Maudet, C.; Transy, C.; Margottin-Goguet, F. Limelight on two HIV/SIV accessory proteins in macrophage infection: Is Vpx overshadowing Vpr? Retrovirology 2010, 7, 35. [Google Scholar] [CrossRef]
- Sunseri, N.; O'Brien, M.; Bhardwaj, N.; Landau, N.R. Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J. Virol. 2011, 85, 6263–6274. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, R.; Zhu, X.; Stranska, R.; Wu, Y.; Stevenson, M. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe 2009, 6, 68–80. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Duncan, C.J.A.; Sattentau, Q.J. Viral Determinants of HIV-1 Macrophage Tropism. Viruses 2011, 3, 2255-2279. https://doi.org/10.3390/v3112255
Duncan CJA, Sattentau QJ. Viral Determinants of HIV-1 Macrophage Tropism. Viruses. 2011; 3(11):2255-2279. https://doi.org/10.3390/v3112255
Chicago/Turabian StyleDuncan, Christopher J. A., and Quentin J. Sattentau. 2011. "Viral Determinants of HIV-1 Macrophage Tropism" Viruses 3, no. 11: 2255-2279. https://doi.org/10.3390/v3112255
APA StyleDuncan, C. J. A., & Sattentau, Q. J. (2011). Viral Determinants of HIV-1 Macrophage Tropism. Viruses, 3(11), 2255-2279. https://doi.org/10.3390/v3112255