Insertional Oncogenesis by Non-Acute Retroviruses: Implications for Gene Therapy

Abstract
:1. Introduction
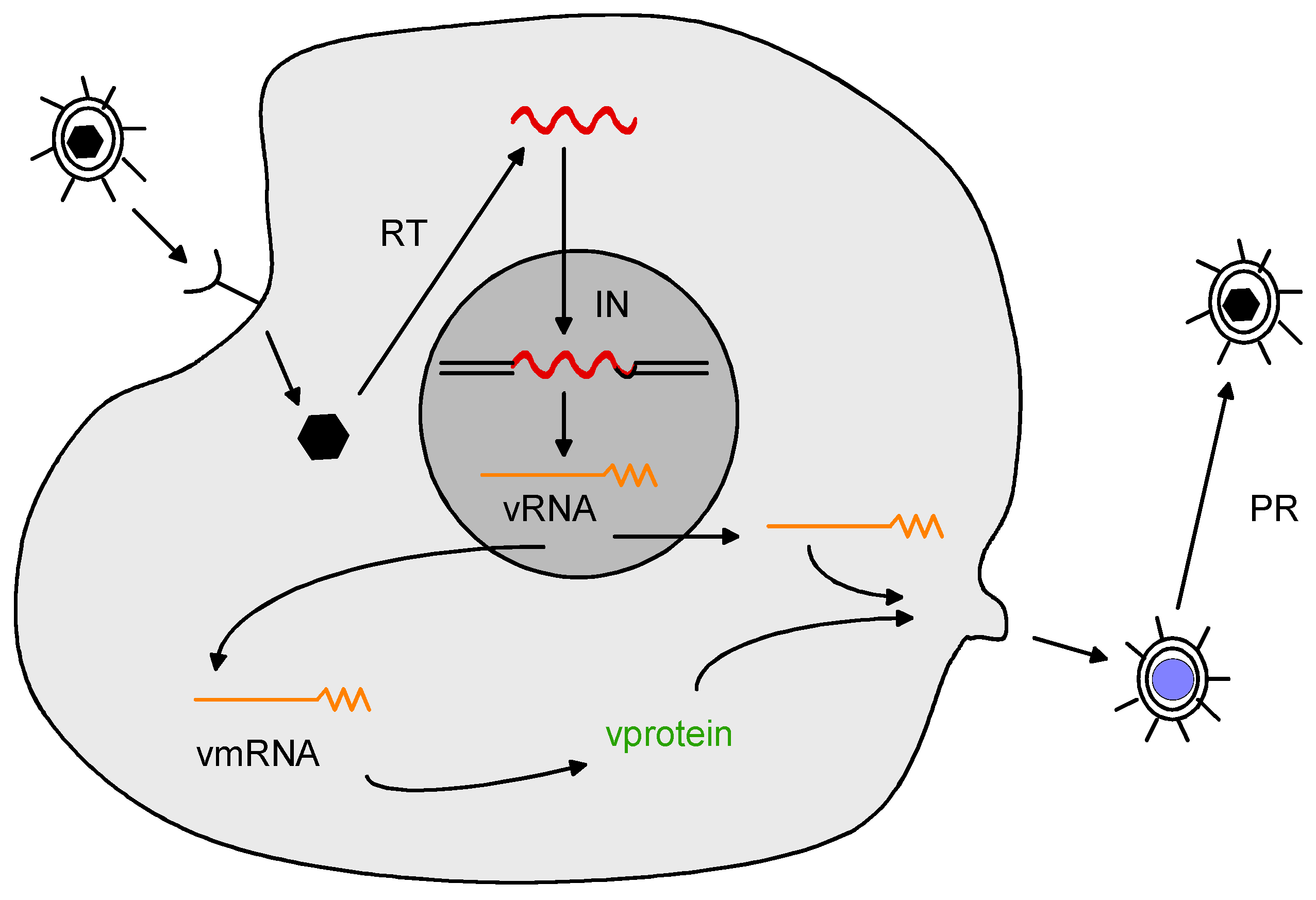
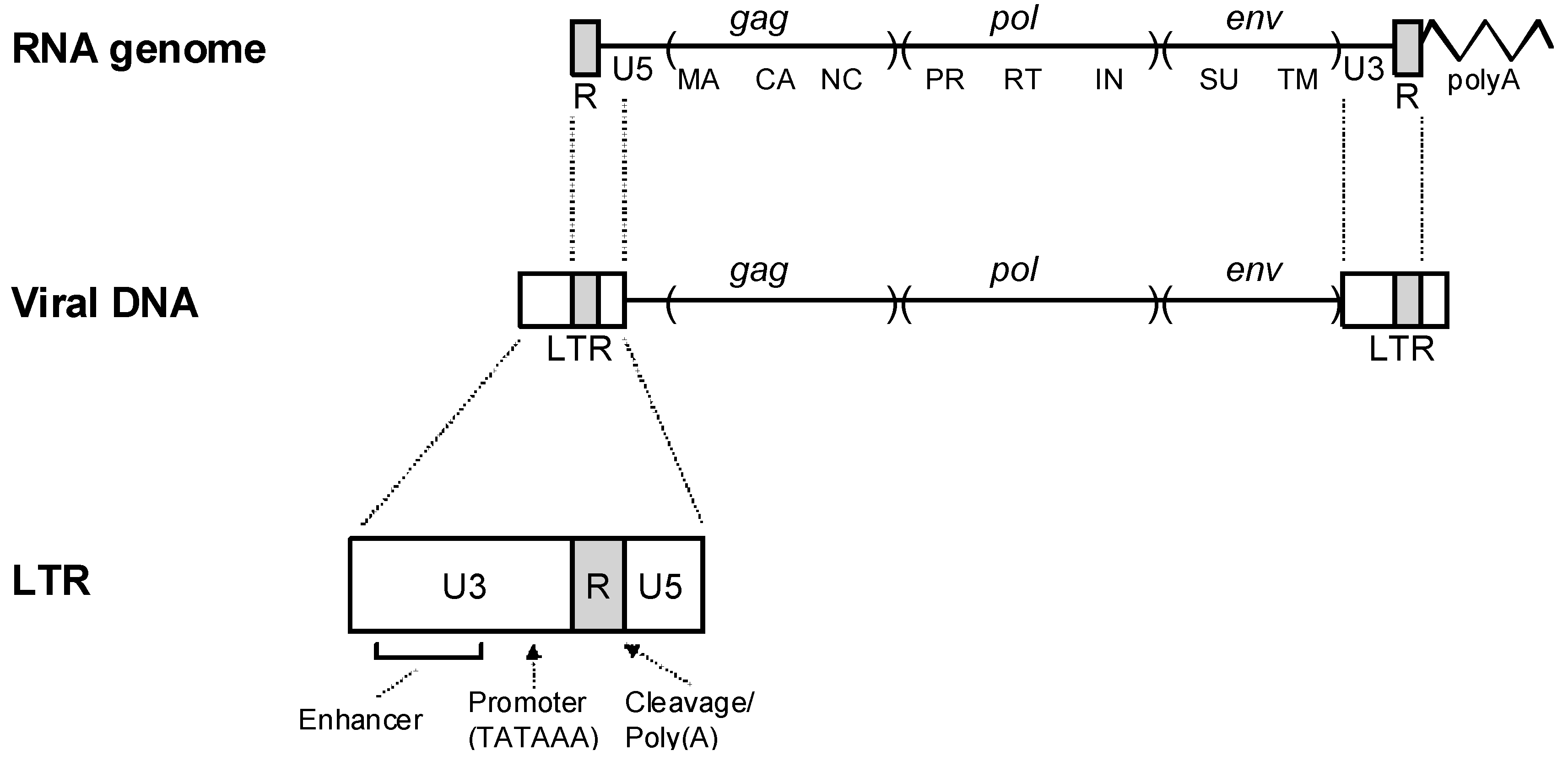
2. Retrovirus Structure and Replication
3. Retroviral Oncogenesis: Acute Transforming vs. Non-Acute Retroviruses
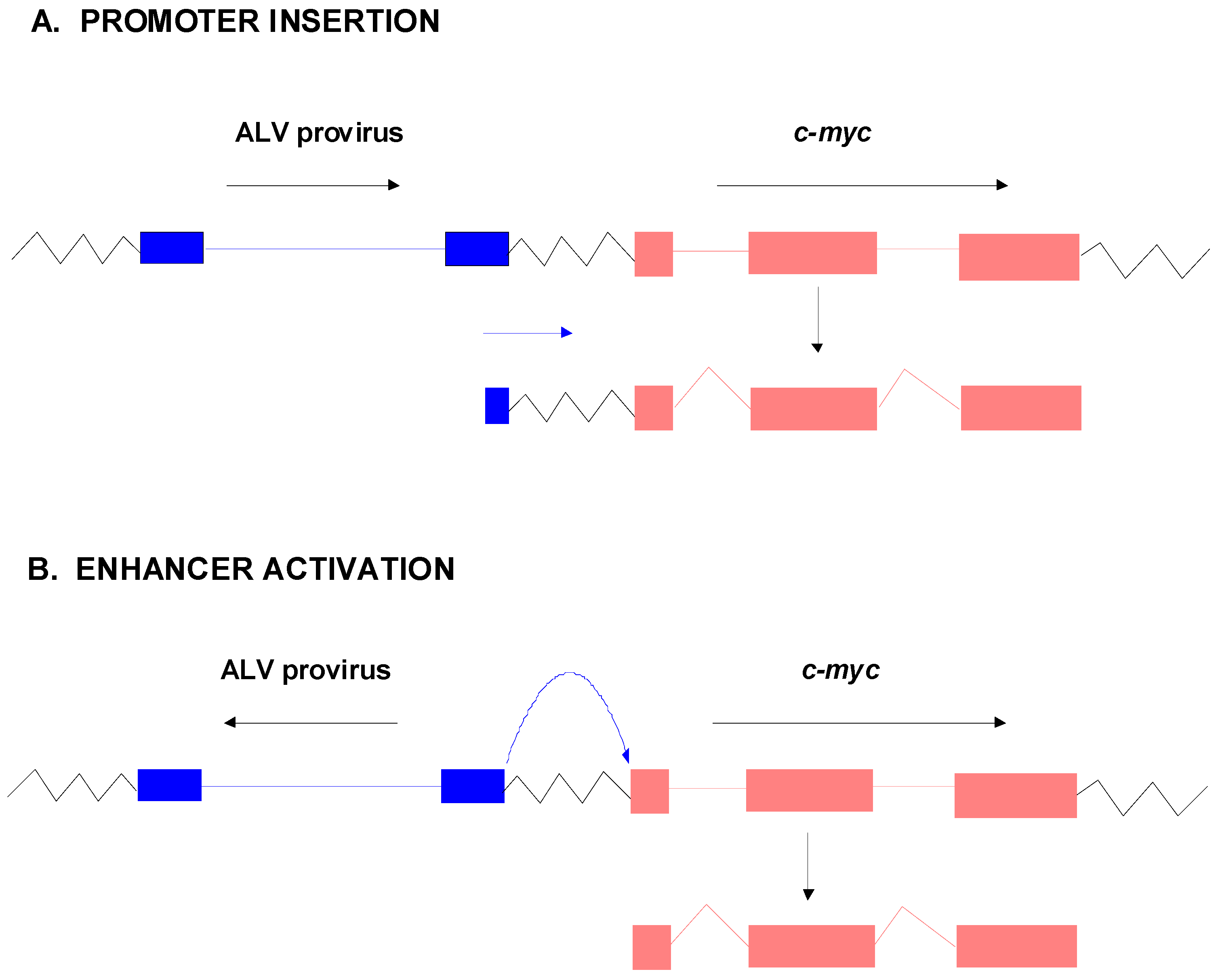
4. Insertional Activation of Proto-Oncogenes
5. Discovery of New Proto-Oncogenes by Studying Insertional Activation
6. LTRs as Determinants of Disease Specificity
7. Cooperation among Activated Proto-Oncogenes
8. Insertional Activation in Multi-Step Carcinogenesis and Tumor Progression
9. Other Mechanisms of Insertional Oncogenesis
10. Insertional Mutagenesis in the Age of Genomics
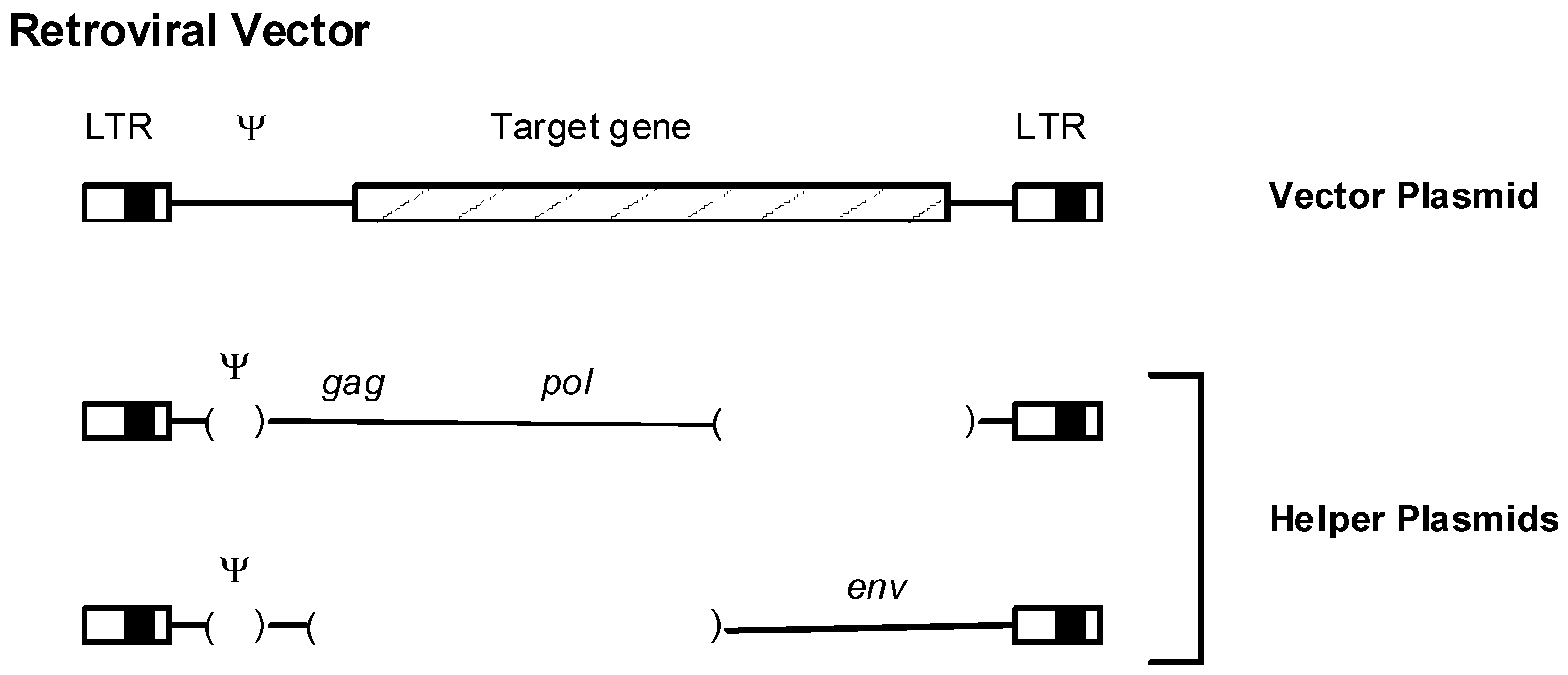
11. Insertional Mutagenesis and Gene Therapy
12. Summary and Perspectives
Acknowledgements
References and Notes
- Urisman, A.; Molinaro, R.J.; Fischer, N.; Plummer, S.J.; Casey, G.; Klein, E.A.; Malathi, K.; Magi-Galluzzi, C.; Tubbs, R.R.; Ganem, D.; Silverman, R.H.; Derisi, J.L. Identification of a Novel Gammaretrovirus in Prostate Tumors of Patients Homozygous for R462Q RNASEL Variant. PLoS Pathog. 2006, 2, e25. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Kim, S.; Hong, S.; Das Gupta, J.; Malathi, K.; Klein, E.A.; Ganem, D.; Derisi, J.L.; Chow, S.A.; Silverman, R.H. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Gray, E.R.; Gall, A.; Katzourakis, A.; Tan, C.P.; Houldcroft, C.J.; McLaren, S.; Pillay, D.; Futreal, A.; Garson, J.A.; Pybus, O.G.; Kellam, P.; Towers, G.J. Disease-associated XMRV sequences are consistent with laboratory contamination. Retrovirology 2010, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Schlaberg, R.; Choe, D.J.; Brown, K.R.; Thaker, H.M.; Singh, I.R. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16351–16356. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Hellwinkel, O.; Schulz, C.; Chun, F.K.; Huland, H.; Aepfelbacher, M.; Schlomm, T. Prevalence of human gammaretrovirus XMRV in sporadic prostate cancer. J. Clin. Virol. 2008, 43, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, V.C.; Ruscetti, F.W.; Das Gupta, J.; Pfost, M.A.; Hagen, K.S.; Peterson, D.L.; Ruscetti, S.K.; Bagni, R.K.; Petrow-Sadowski, C.; Gold, B.; Dean, M.; Silverman, R.H.; Mikovits, J.A. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 2009, 326, 585–589. [Google Scholar] [CrossRef]
- Aloia, A.L.; Sfanos, K.S.; Isaacs, W.B.; Zheng, Q.; Maldarelli, F.; De Marzo, A.M.; Rein, A. XMRV: A New Virus in Prostate Cancer? Cancer Res. 2010, 70, 10028–10033. [Google Scholar] [CrossRef]
- Lo, S.C.; Pripuzova, N.; Li, B.; Komaroff, A.L.; Hung, G.C.; Wang, R.; Alter, H.J. Detection of MLV-related virus gene sequences in blood of patients with chronic fatigue syndrome and healthy blood donors. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15874–15879. [Google Scholar] [CrossRef]
- Groom, H.C.; Boucherit, V.C.; Makinson, K.; Randal, E.; Baptista, S.; Hagan, S.; Gow, J.W.; Mattes, F.M.; Breuer, J.; Kerr, J.R.; Stoye, J.P.; Bishop, K.N. Absence of xenotropic murine leukaemia virus-related virus in UK patients with chronic fatigue syndrome. Retrovirology 2010, 7, 10. [Google Scholar] [CrossRef]
- Smith, R.A. Contamination of clinical specimens with MLV-encoding nucleic acids: implications for XMRV and other candidate human retroviruses. Retrovirology 2010, 7, 112. [Google Scholar] [CrossRef]
- Coffin, J.; Hughes, S.; Varmus, H. Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.; Jolicoeur, P. Retroviral pathogenesis. In Retroviruses; Coffin, J., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; pp. 475–585. [Google Scholar]
- Duesberg, P.H.; Vogt, P.K. Differences between the ribonucleic acids of transforming and nontransforming avian tumor viruses. Proc. Natl. Acad. Sci. U. S. A. 1970, 67, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Rubin, H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology 1958, 6, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Brugge, J.S.; Erikson, R.L. Identification of a transformation-specific antigen induced by an avian sarcoma virus. Nature 1977, 269, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Collett, M.S.; Erikson, R.L. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 2021–2024. [Google Scholar] [CrossRef] [PubMed]
- Collett, M.S.; Purchio, A.F.; Erikson, R.L. Avian sarcoma virus-transforming protein, pp60src shows protein kinase activity specific for tyrosine. Nature 1980, 285, 167–169. [Google Scholar] [CrossRef]
- Hunter, T.; Sefton, B.M. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 1311–1315. [Google Scholar] [CrossRef]
- Sheiness, D.; Fanshier, L.; Bishop, J.M. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J. Virol. 1978, 28, 600–610. [Google Scholar] [CrossRef]
- Graf, T.; Beug, H. Role of the v-erbA and v-erbB oncogenes of avian erythroblastosis virus in erythroid cell transformation. Cell 1983, 34, 7–9. [Google Scholar] [CrossRef]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Shih, T.Y.; Papageorge, A.G.; Stokes, P.E.; Weeks, M.O.; Scolnick, E.M. Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature 1980, 287, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.W.; Defeo, D.; Shih, T.Y.; Gonda, M.A.; Young, H.A.; Tsuchida, N.; Lowy, D.R.; Scolnick, E.M. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 1981, 292, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Willingham, M.C.; Pastan, I.; Shih, T.Y.; Scolnick, E.M. Localization of the src gene product of the Harvey strain of MSV to plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell 1980, 19, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Huleihel, M.; Goldsborough, M.; Cleveland, J.; Gunnell, M.; Bonner, T.; Rapp, U.R. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell Biol. 1986, 6, 2655–2662. [Google Scholar]
- Rapp, U.R.; Goldsborough, M.D.; Mark, G.E.; Bonner, T.I.; Groffen, J.; Reynolds, F.H., Jr.; Stephenson, J.R. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 4218–4222. [Google Scholar] [CrossRef]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J.M. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef]
- Alitalo, K.; Ramsay, G.; Bishop, J.M.; Pfeifer, S.O.; Colby, W.W.; Levinson, A.D. Identification of nuclear proteins encoded by viral and cellular myc oncogenes. Nature 1983, 306, 274–277. [Google Scholar] [CrossRef]
- Van Beveren, C.; van Straaten, F.; Curran, T.; Muller, R.; Verma, I.M. Analysis of FBJ-MuSV provirus and c-fos (mouse) gene reveals that viral and cellular fos gene products have different carboxy termini. Cell 1983, 32, 1241–1255. [Google Scholar] [CrossRef]
- van Straaten, F.; Muller, R.; Curran, T.; Van Beveren, C.; Verma, I.M. Complete nucleotide sequence of a human c-onc gene: deduced amino acid sequence of the human c-fos protein. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 3183–3187. [Google Scholar] [CrossRef]
- Bohmann, D.; Bos, T.J.; Admon, A.; Nishimura, T.; Vogt, P.K.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392. [Google Scholar] [CrossRef]
- Maki, Y.; Bos, T.J.; Davis, C.; Starbuck, M.; Vogt, P.K. Avian sarcoma virus 17 carries the jun oncogene. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 2848–2852. [Google Scholar] [CrossRef]
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [PubMed]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Tabin, C.J.; Bradley, S.M.; Bargmann, C.I.; Weinberg, R.A.; Papageorge, A.G.; Scolnick, E.M.; Dhar, R.; Lowy, D.R.; Chang, E.H. Mechanism of activation of a human oncogene. Nature 1982, 300, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.P.; Reynolds, R.K.; Santos, E.; Barbacid, M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 1982, 300, 149–152. [Google Scholar] [CrossRef]
- Alitalo, K.; Schwab, M.; Lin, C.C.; Varmus, H.E.; Bishop, J.M. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 1707–1711. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Varmus, H.E.; Bishop, J.M.; George, D. A cellular oncogene (c-Ki-ras) is amplified, overexpressed, and located within karyotypic abnormalities in mouse adrenocortical tumour cells. Nature 1983, 303, 497–501. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Martinotti, S.; Gallo, R.C.; Erikson, J.; Croce, C.M. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science 1983, 219, 963–967. [Google Scholar] [CrossRef]
- Marcu, K.B.; Harris, L.J.; Stanton, L.W.; Erikson, J.; Watt, R.; Croce, C.M. Transcriptionally active c-myc oncogene is contained within NIARD, a DNA sequence associated with chromosome translocations in B-cell neoplasia. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 519–523. [Google Scholar] [CrossRef]
- Nowell, P.; Finan, J.; Dalla-Favera, R.; Gallo, R.C.; ar-Rushdi, A.; Romanczuk, H.; Selden, J.R.; Emanuel, B.S.; Rovera, G.; Croce, C.M. Association of amplified oncogene c-myc with an abnormally banded chromosome 8 in a human leukaemia cell line. Nature 1983, 306, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Hayward, W.S.; Neel, B.G.; Astrin, S.M. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 1981, 290, 475–480. [Google Scholar] [CrossRef]
- Payne, G.S.; Bishop, J.M.; Varmus, H.E. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphomas. Nature 1982, 295, 209–214. [Google Scholar] [CrossRef]
- Selten, G.; Cuypers, H.T.; Zijlstra, M.; Melief, C.; Berns, A. Involvement of c-myc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of activation. EMBO J. 1984, 3, 3215–3222. [Google Scholar] [CrossRef]
- Steffen, D. Proviruses are adjacent to c-myc in some murine leukemia virus-induced lymphomas. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Fung, Y.K.; Lewis, W.G.; Crittenden, L.B.; Kung, H.J. Activation of the cellular oncogene c-erbB by LTR insertion: molecular basis for induction of erythroblastosis by avian leukosis virus. Cell 1983, 33, 357–368. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Maroney, P.A.; Goodwin, R.G.; Rottman, F.M.; Crittenden, L.B.; Raines, M.A.; Kung, H.J. c-erbB activation in ALV-induced erythroblastosis: novel RNA processing and promoter insertion result in expression of an amino-truncated EGF receptor. Cell 1985, 41, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Raines, M.A.; Lewis, W.G.; Crittenden, L.B.; Kung, H.J. c-erbB activation in avian leukosis virus-induced erythroblastosis: clustered integration sites and the arrangement of provirus in the c-erbB alleles. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 2287–2291. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Peters, G.; Brookes, S.; Smith, R.; Dickson, C. Tumorigenesis by mouse mammary tumor virus: evidence for a common region for provirus integration in mammary tumors. Cell 1983, 33, 369–377. [Google Scholar] [CrossRef]
- Dickson, C.; Smith, R.; Brookes, S.; Peters, G. Tumorigenesis by mouse mammary tumor virus: proviral activation of a cellular gene in the common integration region int-2. Cell 1984, 37, 529–536. [Google Scholar] [CrossRef]
- Gallahan, D.; Kozak, C.; Callahan, R. A new common integration region (int-3) for mouse mammary tumor virus on mouse chromosome 17. J. Virol. 1987, 61, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. WNT and FGF gene clusters (review). Int. J. Oncol. 2002, 21, 1269–1273. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, H.T.; Selten, G.; Quint, W.; Zijlstra, M.; Maandag, E.R.; Boelens, W.; van Wezenbeek, P.; Melief, C.; Berns, A. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell 1984, 37, 141–150. [Google Scholar] [CrossRef]
- Breuer, M.L.; Cuypers, H.T.; Berns, A. Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas. EMBO J. 1989, 8, 743–748. [Google Scholar] [CrossRef]
- Jolicoeur, P.; Villeneuve, L.; Rassart, E.; Kozak, C. Mouse chromosomal mapping of a murine leukemia virus integration region (Mis-1) first identified in rat thymic leukemia. J. Virol. 1985, 56, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Gachelin, F.; Tavitian, A.; Tambourin, P. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature 1988, 331, 277–280. [Google Scholar] [CrossRef]
- Paul, R.; Schuetze, S.; Kozak, S.L.; Kabat, D. A common site for immortalizing proviral integrations in Friend erythroleukemia: molecular cloning and characterization. J. Virol. 1989, 63, 4958–4961. [Google Scholar] [CrossRef]
- Ben-David, Y.; Giddens, E.B.; Bernstein, A. Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 1332–1336. [Google Scholar] [CrossRef]
- Copeland, N.G.; Jenkins, N.A. Myeloid leukemia: Disease genes and mouse models. Prog. Exp. Tumor Res. 1999, 35, 53–63. [Google Scholar]
- Copeland, N.G.; Jenkins, N.A. Retroviral integration in murine myeloid tumors to identify Evi-1, a novel locus encoding a zinc-finger protein. Adv. Cancer Res. 1990, 54, 141–157. [Google Scholar] [PubMed]
- Mouse Retroviral Tagged Cancer Gene Database. Available online: http://RTCGD.ncifcrf.gov (accessed on 1 March 2011).
- Akagi, K.; Suzuki, T.; Stephens, R.M.; Jenkins, N.A.; Copeland, N.G. RTCGD: Retroviral tagged cancer gene database. Nucleic Acids Res. 2004, 32, D523–D527. [Google Scholar] [CrossRef]
- Holland, C.A.; Thomas, C.Y.; Chattopadhyay, S.K.; Koehne, C.; O’Donnell, P.V. Influence of enhancer sequences on thymotropism and leukemogenicity of mink cell focus-forming viruses. J. Virol. 1989, 63, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Lenz, J.; Celander, D.; Crowther, R.L.; Patarca, R.; Perkins, D.W.; Haseltine, W.A. Determination of the leukaemogenicity of a murine retrovirus by sequences within the long terminal repeat. Nature 1984, 308, 467–470. [Google Scholar] [CrossRef]
- Chatis, P.A.; Holland, C.A.; Silver, J.E.; Frederickson, T.N.; Hopkins, N.; Hartley, J.W. A 3’ end fragment encompassing the transcriptional enhancers of nondefective Friend virus confers erythroleukemogenicity on Moloney leukemia virus. J. Virol. 1984, 52, 248–254. [Google Scholar] [CrossRef]
- Li, Y.; Golemis, E.; Hartley, J.W.; Hopkins, N. Disease specificity of nondefective Friend and Moloney murine leukemia viruses is controlled by a small number of nucleotides. J. Virol. 1987, 61, 693–700. [Google Scholar] [CrossRef]
- Short, M.K.; Okenquist, S.A.; Lenz, J. Correlation of leukemogenic potential of murine retroviruses with transcriptional tissue preference of the viral long terminal repeats. J. Virol. 1987, 61, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.; Wagenaar, E.; Sluyser, M.; Nusse, R. Activation of int-1 and int-2 mammary oncogenes in hormone-dependent and -independent mammary tumors of GR mice. J. Virol. 1987, 61, 1073–1078. [Google Scholar] [CrossRef]
- Cuypers, H.T.; Selten, G.C.; Zijlstra, M.; de Goede, R.E.; Melief, C.J.; Berns, A.J. Tumor progression in murine leukemia virus-induced T-cell lymphomas: monitoring clonal selections with viral and cellular probes. J. Virol. 1986, 60, 230–241. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Krimpenfort, P.; Domen, J.; Saris, C.; Radaszkiewicz, T.; Berns, A. Predisposition to lymphomagenesis in pim-1 transgenic mice: Cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989, 56, 673–682. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- van der Lugt, N.M.; Domen, J.; Verhoeven, E.; Linders, K.; van der Gulden, H.; Allen, J.; Berns, A. Proviral tagging in E mu-myc transgenic mice lacking the Pim-1 proto-oncogene leads to compensatory activation of Pim-2. EMBO J. 1995, 14, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Mikkers, H.; Allen, J.; Knipscheer, P.; Romeijn, L.; Hart, A.; Vink, E.; Berns, A. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat. Genet. 2002, 32, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Girard, L.; Hanna, Z.; Beaulieu, N.; Hoemann, C.D.; Simard, C.; Kozak, C.A.; Jolicoeur, P. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes Dev. 1996, 10, 1930–1944. [Google Scholar] [CrossRef]
- Stewart, M.; Cameron, E.; Campbell, M.; McFarlane, R.; Toth, S.; Lang, K.; Onions, D.; Neil, J.C. Conditional expression and oncogenicity of c-myc linked to a CD2 gene dominant control region. Int. J. Cancer 1993, 53, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.; Terry, A.; Hu, M.; O’Hara, M.; Blyth, K.; Baxter, E.; Cameron, E.; Onions, D.E.; Neil, J.C. Proviral insertions induce the expression of bone-specific isoforms of PEBP2alphaA (CBFA1): evidence for a new myc collaborating oncogene. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8646–8651. [Google Scholar] [CrossRef]
- Stewart, M.; Mackay, N.; Hanlon, L.; Blyth, K.; Scobie, L.; Cameron, E.; Neil, J.C. Insertional mutagenesis reveals progression genes and checkpoints in MYC/Runx2 lymphomas. Cancer Res. 2007, 67, 5126–5133. [Google Scholar] [CrossRef]
- Kool, J.; Uren, A.G.; Martins, C.P.; Sie, D.; de Ridder, J.; Turner, G.; van Uitert, M.; Matentzoglu, K.; Lagcher, W.; Krimpenfort, P.; et al. Insertional mutagenesis in mice deficient for p15Ink4b, p16Ink4a, p21Cip1, and p27Kip1 reveals cancer gene interactions and correlations with tumor phenotypes. Cancer Res. 2010, 70, 520–531. [Google Scholar] [CrossRef]
- Tanaka, M.; Jin, G.; Yamazaki, Y.; Takahara, T.; Takuwa, M.; Nakamura, T. Identification of candidate cooperative genes of the Apc mutation in transformation of the colon epithelial cell by retroviral insertional mutagenesis. Cancer Sci. 2008, 99, 979–985. [Google Scholar] [CrossRef]
- Savard, P.; DesGroseillers, L.; Rassart, E.; Poirier, Y.; Jolicoeur, P. Important role of the long terminal repeat of the helper Moloney murine leukemia virus in Abelson virus-induced lymphoma. J. Virol. 1987, 61, 3266–3275. [Google Scholar] [CrossRef]
- Poirier, Y.; Kozak, C.; Jolicoeur, P. Identification of a common helper provirus integration site in Abelson murine leukemia virus-induced lymphoma DNA. J. Virol. 1988, 62, 3985–3992. [Google Scholar] [CrossRef]
- Jiang, X.; Hanna, Z.; Kaouass, M.; Girard, L.; Jolicoeur, P. Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J. Virol. 2002, 76, 9046–9059. [Google Scholar] [CrossRef]
- Bear, S.E.; Bellacosa, A.; Lazo, P.A.; Jenkins, N.A.; Copeland, N.G.; Hanson, C.; Levan, G.; Tsichlis, P.N. Provirus insertion in Tpl-1, an Ets-1-related oncogene, is associated with tumor progression in Moloney murine leukemia virus-induced rat thymic lymphomas. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7495–7499. [Google Scholar] [CrossRef] [PubMed]
- Patriotis, C.; Makris, A.; Bear, S.E.; Tsichlis, P.N. Tumor progression locus 2 (Tpl-2) encodes a protein kinase involved in the progression of rodent T-cell lymphomas and in T-cell activation. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2251–2255. [Google Scholar] [CrossRef] [PubMed]
- Flubacher, M.M.; Bear, S.E.; Tsichlis, P.N. Replacement of interleukin-2 (IL-2)-generated mitogenic signals by a mink cell focus-forming (MCF) or xenotropic virus-induced IL-9-dependent autocrine loop: Implications for MCF virus-induced leukemogenesis. J. Virol. 1994, 68, 7709–7716. [Google Scholar] [CrossRef]
- Gilks, C.B.; Bear, S.E.; Grimes, H.L.; Tsichlis, P.N. Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol. Cell Biol. 1993, 13, 1759–1768. [Google Scholar]
- Heard, J.M.; Fichelson, S.; Sola, B.; Martial, M.A.; Varet, B.; Levy, J.P. Multistep virus-induced leukemogenesis in vitro: description of a model specifying three steps within the myeloblastic malignant process. Mol. Cell Biol. 1984, 4, 216–220. [Google Scholar]
- Sola, B.; Fichelson, S.; Bordereaux, D.; Tambourin, P.E.; Gisselbrecht, S. fim-1 and fim-2: Two new integration regions of Friend murine leukemia virus in myeloblastic leukemias. J. Virol. 1986, 60, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Gisselbrecht, S.; Fichelson, S.; Sola, B.; Bordereaux, D.; Hampe, A.; Andre, C.; Galibert, F.; Tambourin, P. Frequent c-fms activation by proviral insertion in mouse myeloblastic leukaemias. Nature 1987, 329, 259–261. [Google Scholar] [CrossRef]
- Ben-David, Y.; Lavigueur, A.; Cheong, G.Y.; Bernstein, A. Insertional inactivation of the p53 gene during friend leukemia: a new strategy for identifying tumor suppressor genes. New Biol. 1990, 2, 1015–1023. [Google Scholar] [PubMed]
- Mowat, M.; Cheng, A.; Kimura, N.; Bernstein, A.; Benchimol, S. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 1985, 314, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Largaespada, D.A.; Shaughnessy, J.D., Jr.; Jenkins, N.A.; Copeland, N.G. Retroviral integration at the Evi-2 locus in BXH-2 myeloid leukemia cell lines disrupts Nf1 expression without changes in steady-state Ras-GTP levels. J. Virol. 1995, 69, 5095–5102. [Google Scholar] [CrossRef]
- Nason-Burchenal, K.; Wolff, L. Activation of c-myb is an early bone-marrow event in a murine model for acute promonocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 1619–1623. [Google Scholar] [CrossRef]
- Shen-Ong, G.L.; Wolff, L. Moloney murine leukemia virus-induced myeloid tumors in adult BALB/c mice: Requirement of c-myb activation but lack of v-abl involvement. J. Virol. 1987, 61, 3721–3725. [Google Scholar] [CrossRef]
- Wolff, L.; Mushinski, J.F.; Shen-Ong, G.L.; Morse, H.C., 3rd. A chronic inflammatory response. Its role in supporting the development of c-myb and c-myc related promonocytic and monocytic tumors in BALB/c mice. J. Immunol. 1988, 141, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Belli, B.; Wolff, L.; Nazarov, V.; Fan, H. Proviral activation of the c-myb proto-oncogene is detectable in preleukemic mice infected neonatally with Moloney murine leukemia virus but not in resulting end stage T lymphomas. J. Virol. 1995, 69, 5138–5141. [Google Scholar] [CrossRef]
- Clurman, B.E.; Hayward, W.S. Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol. Cell Biol. 1989, 9, 2657–2664. [Google Scholar]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar] [CrossRef]
- Bolisetty, M.T.; Dy, G.; Tam, W.; Beemon, K.L. Reticuloendotheliosis virus strain T induces miR-155, which targets JARID2 and promotes cell survival. J. Virol. 2009, 83, 12009–12017. [Google Scholar] [CrossRef]
- Landais, S.; Landry, S.; Legault, P.; Rassart, E. Oncogenic potential of the miR-106–363 cluster and its implication in human T-cell leukemia. Cancer Res. 2007, 67, 5699–5707. [Google Scholar] [CrossRef] [PubMed]
- Lum, A.M.; Wang, B.B.; Li, L.; Channa, N.; Bartha, G.; Wabl, M. Retroviral activation of the mir-106a microRNA cistron in T lymphoma. Retrovirology 2007, 4, 5. [Google Scholar] [CrossRef]
- Dabrowska, M.J.; Dybkaer, K.; Johnsen, H.E.; Wang, B.; Wabl, M.; Pedersen, F.S. Loss of MicroRNA targets in the 3’ untranslated region as a mechanism of retroviral insertional activation of growth factor independence 1. J. Virol. 2009, 83, 8051–8061. [Google Scholar] [CrossRef]
- Li, J.; Shen, H.; Himmel, K.L.; Dupuy, A.J.; Largaespada, D.A.; Nakamura, T.; Shaughnessy, J.D., Jr.; Jenkins, N.A.; Copeland, N.G. Leukaemia disease genes: Large-scale cloning and pathway predictions. Nat. Genet. 1999, 23, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; Turner, G.; Trubetskoy, A.; Verhoeven, E.; Wientjens, E.; Hulsman, D.; Russell, R.; DePinho, R.A.; Lenz, J.; van Lohuizen, M. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 2002, 32, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Martins, C.P.; Bronkhorst, Y.; Randel, E.; Berns, A.; Fero, M.; Clurman, B.E. Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and high-throughput insertion site analysis. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11293–11298. [Google Scholar] [CrossRef]
- Suzuki, T.; Shen, H.; Akagi, K.; Morse, H.C.; Malley, J.D.; Naiman, D.Q.; Jenkins, N.A.; Copeland, N.G. New genes involved in cancer identified by retroviral tagging. Nat. Genet. 2002, 32, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.; Kool, J.; Uren, A.G.; de Ridder, J.; Wessels, L.; Jonkers, J.; Bignell, G.R.; Butler, A.; Rust, A.G.; Brosch, M.; et al. Novel candidate cancer genes identified by a large-scale cross-species comparative oncogenomics approach. Cancer Res. 2010, 70, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Makunin, I.V.; Pheasant, M.; Simons, C.; Mattick, J.S. Orthologous microRNA genes are located in cancer-associated genomic regions in human and mouse. PLoS ONE 2007, 2, e1133. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D. Retroviral vectors. Curr. Top. Microbiol. Immunol. 1992, 158, 1–24. [Google Scholar] [PubMed]
- Matrai, J.; Chuah, M.K.; VandenDriessche, T. Recent advances in lentiviral vector development and applications. Mol. Ther. 2010, 18, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef]
- Ferrua, F.; Brigida, I.; Aiuti, A. Update on gene therapy for adenosine deaminase-deficient severe combined immunodeficiency. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; Bousso, P.; Deist, F.L.; Fischer, A. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; Fischer, A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Rabbitts, T.H.; Axelson, H.; Forster, A.; Grutz, G.; Lavenir, I.; Larson, R.; Osada, H.; Valge-Archer, V.; Wadman, I.; Warren, A. Chromosomal translocations and leukaemia: A role for LMO2 in T cell acute leukaemia, in transcription and in erythropoiesis. Leukemia 1997, 11, 271–272. [Google Scholar]
- Krause, D. Gene Therapy for Wiskott-Aldrich Syndrome: Benefits and Risks. Hematologist. 1 March 2011. Available online: http://www.hematology.org/Publications/Hematologist/2011/6487.aspx (accessed on 17 March 2011).
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Diez, I.A.; Dewey, R.A.; Bohm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 3194–3198. [Google Scholar] [CrossRef]
- Chung, J.H.; Whiteley, M.; Felsenfeld, G. A 5’ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 1993, 74, 505–514. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M. Oncogenic HMGA2: Short or small? Genes Dev. 2007, 21, 1005–1009. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VIRUS | DISEASE | CIS or PROTO-ONCOGENE |
|---|---|---|
| Moloney MuLV | T-lymphoma | |
| In mice: c-myc, pim-1, pvt-1/mis-1/mlvi-1, lck, pim-2a, n-myca, bmi-1a, frat-1a, pal-1/gfi-1a | ||
| In rats: c-myc, pvt-1/mis-1/mlvi-1, mlvi-2, mlvi-3, mlvi-4, dsi-1, lck, tpl-1/ets-1a, tpl-2a, gfi-1/pal-1a, gfi-2/IL-9R | ||
| Myeloid leukemia | c-myb, mml-1 | |
| AKR MuLV/Gross Virus; SL3-3 MuLV | T-lymphoma | c-myc, gin-1, n-ras |
| RadLV (Radiation leukemia virus) | T-lymphoma | c-myc, pim-1, vin-1/cyclinD2, notch1, kis-1, kis-2 |
| Friend MuLV | Erythroleukemia | fli-1, fre-2 |
| Myeloid leukemia | fis-1, fim-1, evi-1/fim-3, c-fms/fim-2 | |
| Endogenous MuLV (AKXD, BXH-2 recombinant inbred mice) | Myeloid leukemia | evi-1/fim-3, evi-2, meis-1 and others1 |
| B-lymphoma | evi-3 and others1 | |
| Abelson MuLV (contains v-abl oncogene) | B-lymphoma | ahi-1, ahi-2 (M-MuLV helper inserted) |
| Friend SSFV (SFFV gp52c is an oncogene) | Erythroleukemia | Spi-1, p53b |
© 2011 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.; Johnson, C. Insertional Oncogenesis by Non-Acute Retroviruses: Implications for Gene Therapy. Viruses 2011, 3, 398-422. https://doi.org/10.3390/v3040398
Fan H, Johnson C. Insertional Oncogenesis by Non-Acute Retroviruses: Implications for Gene Therapy. Viruses. 2011; 3(4):398-422. https://doi.org/10.3390/v3040398
Chicago/Turabian StyleFan, Hung, and Chassidy Johnson. 2011. "Insertional Oncogenesis by Non-Acute Retroviruses: Implications for Gene Therapy" Viruses 3, no. 4: 398-422. https://doi.org/10.3390/v3040398
APA StyleFan, H., & Johnson, C. (2011). Insertional Oncogenesis by Non-Acute Retroviruses: Implications for Gene Therapy. Viruses, 3(4), 398-422. https://doi.org/10.3390/v3040398