Unconventional Use of LC3 by Coronaviruses through the Alleged Subversion of the ERAD Tuning Pathway

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CoV Life Cycle
3. The ER Origin of the CoV-Induced DMVs
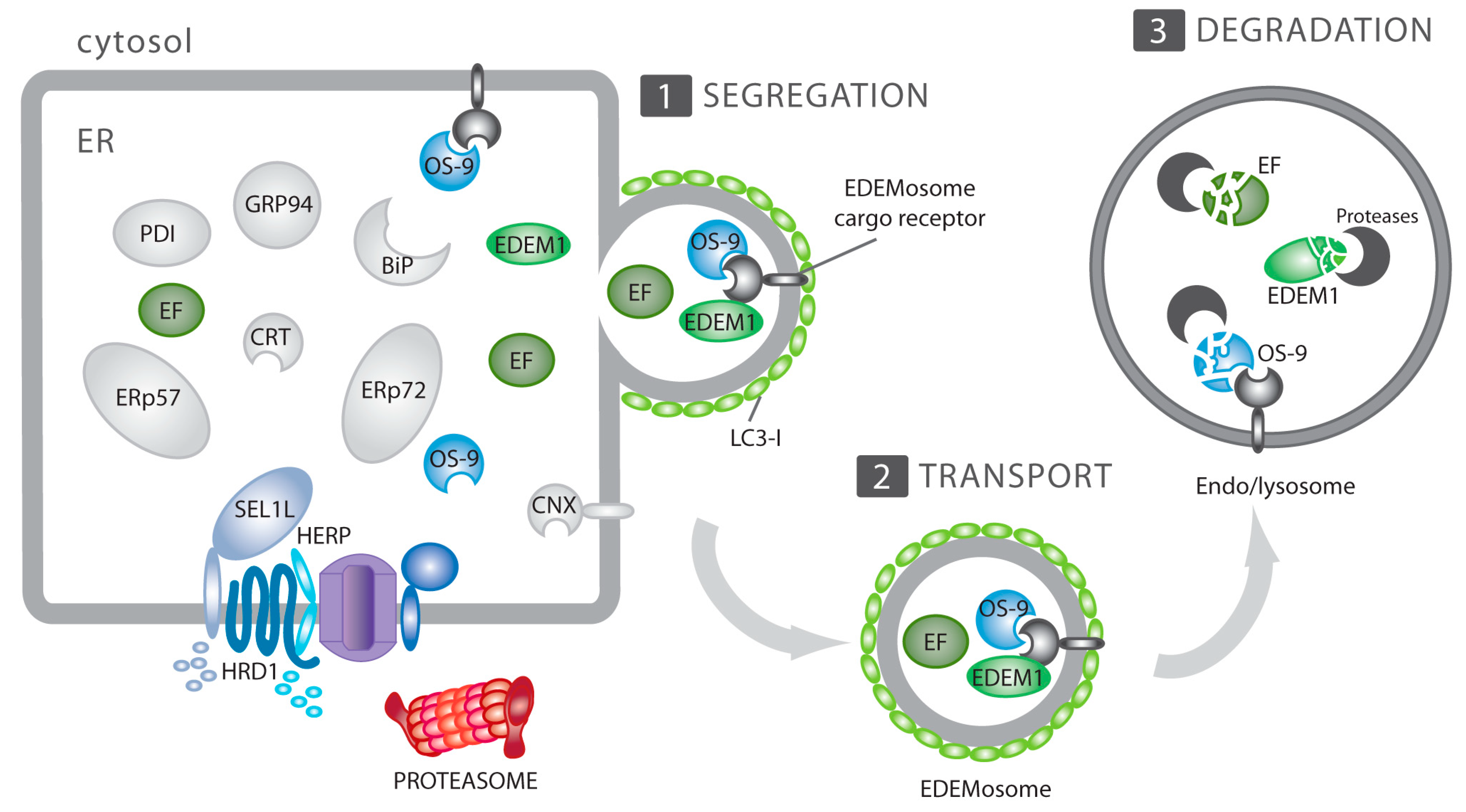
4. ERAD, ERAD Tuning and its Subversion by CoV
5. Unanswered Questions
6. Perspectives
Acknowledgments
References and Notes
- International Commitee on Virus Taxonomy. http://www.ictvonline.org (assessed on 1 September 2011).
- Kahn, J.S.; McIntosh, K. History and recent advances in coronavirus discovery. Pediatr. Infect. Dis. J. 2005, 24, S223–227. [Google Scholar] [CrossRef]
- Garbino, J.; Crespo, S.; Aubert, J.D.; Rochat, T.; Ninet, B.; Deffernez, C.; Wunderli, W.; Pache, J.C.; Soccal, P.M.; Kaiser, L. A prospective hospital-based study of the clinical impact of non-severe acute respiratory syndrome (Non-SARS)-related human coronavirus infection. Clin. Infect. Dis. 2006, 43, 1009–1015. [Google Scholar] [CrossRef]
- Drosten, C.; Preiser, W.; Gunther, S.; Schmitz, H.; Doerr, H.W. Severe acute respiratory syndrome: identification of the etiological agent. Trends Mol. Med. 2003, 9, 325–327. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–97. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Wevers, B.A.; van der Hoek, L. Recently discovered human coronaviruses. Clin. Lab. Med. 2009, 29, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef]
- Kim, L.; Hayes, J.; Lewis, P.; Parwani, A.V.; Chang, K.O.; Saif, L.J. Molecular characterization and pathogenesis of transmissible gastroenteritis coronavirus (TGEV) and porcine respiratory coronavirus (PRCV) field isolates co-circulating in a swine herd. Arch. Virol. 2000, 145, 1133–1147. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef]
- Hartmann, K.; Ritz, S. Treatment of cats with feline infectious peritonitis. Vet. Immunol. Immunopathol. 2008, 123, 172–175. [Google Scholar] [CrossRef]
- Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; Lutz, H.; et al. Feline infectious peritonitis. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 594–604. [Google Scholar] [CrossRef]
- Andrew, S.E. Feline infectious peritonitis. Vet. Clin. North Am. Small Anim. Pract. 2000, 30, 987–1000. [Google Scholar] [CrossRef]
- Saif, L.J. Bovine respiratory coronavirus. Vet. Clin. North Am. Food Anim. Pract. 2010, 26, 349–364. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef]
- Barcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.; Verkleij, A.; Rottier, P.J.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 582–587. [Google Scholar] [CrossRef]
- Sturman, L.S.; Holmes, K.V.; Behnke, J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J. Virol. 1980, 33, 449–462. [Google Scholar] [CrossRef]
- Niemann, H.; Klenk, H.D. Coronavirus glycoprotein E1, a new type of viral glycoprotein. J. Mol. Biol. 1981, 153, 993–1010. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Li, W.; Radoshitzky, S.R.; Choe, H.; Farzan, M. Severe acute respiratory syndrome coronavirus entry as a target of antiviral therapies. Antivir. Ther. 2007, 12, 639–650. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Eifart, P.; Ludwig, K.; Bottcher, C.; de Haan, C.A.; Rottier, P.J.; Korte, T.; Herrmann, A. Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J. Virol. 2007, 81, 10758–10768. [Google Scholar] [CrossRef]
- Chu, V.C.; McElroy, L.J.; Chu, V.; Bauman, B.E.; Whittaker, G.R. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J. Virol. 2006, 80, 3180–3188. [Google Scholar] [CrossRef]
- Huang, I.C.; Bosch, B.J.; Li, F.; Li, W.; Lee, K.H.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar] [CrossRef]
- Ziebuhr, J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- van Hemert, M.J.; van den Worm, S.H.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Tatti, K.M.; Ksiazek, T.G.; Rollin, P.E.; Comer, J.A.; Lee, W.W.; Rota, P.A.; Bankamp, B.; Bellini, W.J.; Zaki, S.R. Ultrastructural characterization of SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 320–326. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Nidovirus transcription: How to make sense...? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef]
- den Boon, J.A.; Ahlquist, P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Gantier, M.P.; Williams, B.R. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007, 18, 363–371. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar]
- Satija, N.; Lal, S.K. The molecular biology of SARS coronavirus. Ann. N. Y. Acad. Sci. 2007, 1102, 26–38. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Hosting the severe acute respiratory syndrome coronavirus: specific cell factors required for infection. Cell Microbiol. 2006, 8, 1211–1218. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.; Te Velthuis, A.J.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 2010, 84, 833–846. [Google Scholar] [CrossRef]
- Oostra, M.; Te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.; de Haan, C.A. Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef]
- Oostra, M.; Hagemeijer, M.C.; van Gent, M.; Bekker, C.P.; te Lintelo, E.G.; Rottier, P.J.; de Haan, C.A. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 2008, 82, 12392–12405. [Google Scholar] [CrossRef]
- Kanjanahaluethai, A.; Chen, Z.; Jukneliene, D.; Baker, S.C. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 2007, 361, 391–401. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Aebi, M.; Bernasconi, R.; Clerc, S.; Molinari, M. N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. [Google Scholar] [CrossRef]
- Hebert, D.N.; Bernasconi, R.; Molinari, M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010, 21, 526–532. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Mendoza, A.; Mariano, J.M.; Zhou, M.; Kostova, Z.; Chen, B.; Veenstra, T.; Hewitt, S.M.; Helman, L.J.; Khanna, C.; et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat. Med. 2007, 13, 1504–1509. [Google Scholar] [CrossRef]
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef]
- Wu, Y.; Termine, D.J.; Swulius, M.T.; Moremen, K.W.; Sifers, R.N. Human endoplasmic reticulum mannosidase I is subject to regulated proteolysis. J. Biol. Chem. 2007, 282, 4841–4849. [Google Scholar] [CrossRef]
- Termine, D.J.; Moremen, K.W.; Sifers, R.N. The mammalian UPR boosts glycoprotein ERAD by suppressing the proteolytic downregulation of ER mannosidase I. J. Cell Sci. 2009, 122, 976–984. [Google Scholar] [CrossRef]
- Calì, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef]
- Le Fourn, V.; Gaplovska-Kysela, K.; Guhl, B.; Santimaria, R.; Zuber, C.; Roth, J. Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell Mol. Life Sci. 2009, 66, 1434–1445. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. [Google Scholar] [CrossRef]
- Miura, H.; Hashida, K.; Sudo, H.; Awa, Y.; Takarada-Iemata, M.; Kokame, K.; Takahashi, T.; Matsumoto, M.; Kitao, Y.; Hori, O. Deletion of Herp facilitates degradation of cytosolic proteins. Genes Cells 2010, 15, 843–853. [Google Scholar] [CrossRef]
- Hori, O.; Ichinoda, F.; Yamaguchi, A.; Tamatani, T.; Taniguchi, M.; Koyama, Y.; Katayama, T.; Tohyama, M.; Stern, D.M.; Ozawa, K.; et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells 2004, 9, 457–469. [Google Scholar] [CrossRef]
- Mueller, B.; Lilley, B.N.; Ploegh, H.L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006, 175, 261–270. [Google Scholar] [CrossRef]
- Zuber, C.; Cormier, J.H.; Guhl, B.; Santimaria, R.; Hebert, D.N.; Roth, J. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4407–4412. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Yoshimori, T.; Noda, T. Toward unraveling membrane biogenesis in mammalian autophagy. Curr. Opin. Cell Biol. 2008, 20, 401–407. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Sou, Y.S.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W.t. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- de Haan, C.A.; Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2008, 4, 276–279. [Google Scholar] [CrossRef]
- Calì, T.; Vanoni, O.; Molinari, M. The endoplasmic reticulum crossroads for newly synthesized polypeptide chains. Prog. Mol. Biol. Transl. Sci. 2008, 83, 135–179. [Google Scholar]
- de Haan, C.A.; Molinari, M.; Reggiori, F. Autophagy-independent LC3 function in vesicular traffic. Autophagy 2010, 6, 994–996. [Google Scholar] [CrossRef]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 2009, 4, e8342. [Google Scholar] [CrossRef]
- Mann, S.S.; Hammarback, J.A. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem. 1994, 269, 11492–11497. [Google Scholar] [CrossRef]
- Pedrotti, B.; Ulloa, L.; Avila, J.; Islam, K. Characterization of microtubule-associated protein MAP1B: phosphorylation state, light chains, and binding to microtubules. Biochemistry 1996, 35, 3016–3023. [Google Scholar] [CrossRef]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 2009, 84, 431–448. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef]
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006, 281, 36303–36316. [Google Scholar] [CrossRef]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiel, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef]


© 2011 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reggiori, F.; De Haan, C.A.M.; Molinari, M. Unconventional Use of LC3 by Coronaviruses through the Alleged Subversion of the ERAD Tuning Pathway. Viruses 2011, 3, 1610-1623. https://doi.org/10.3390/v3091610
Reggiori F, De Haan CAM, Molinari M. Unconventional Use of LC3 by Coronaviruses through the Alleged Subversion of the ERAD Tuning Pathway. Viruses. 2011; 3(9):1610-1623. https://doi.org/10.3390/v3091610
Chicago/Turabian StyleReggiori, Fulvio, Cornelis A.M. De Haan, and Maurizio Molinari. 2011. "Unconventional Use of LC3 by Coronaviruses through the Alleged Subversion of the ERAD Tuning Pathway" Viruses 3, no. 9: 1610-1623. https://doi.org/10.3390/v3091610
APA StyleReggiori, F., De Haan, C. A. M., & Molinari, M. (2011). Unconventional Use of LC3 by Coronaviruses through the Alleged Subversion of the ERAD Tuning Pathway. Viruses, 3(9), 1610-1623. https://doi.org/10.3390/v3091610