dsRNA-Dependent Protein Kinase PKR and its Role in Stress, Signaling and HCV Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
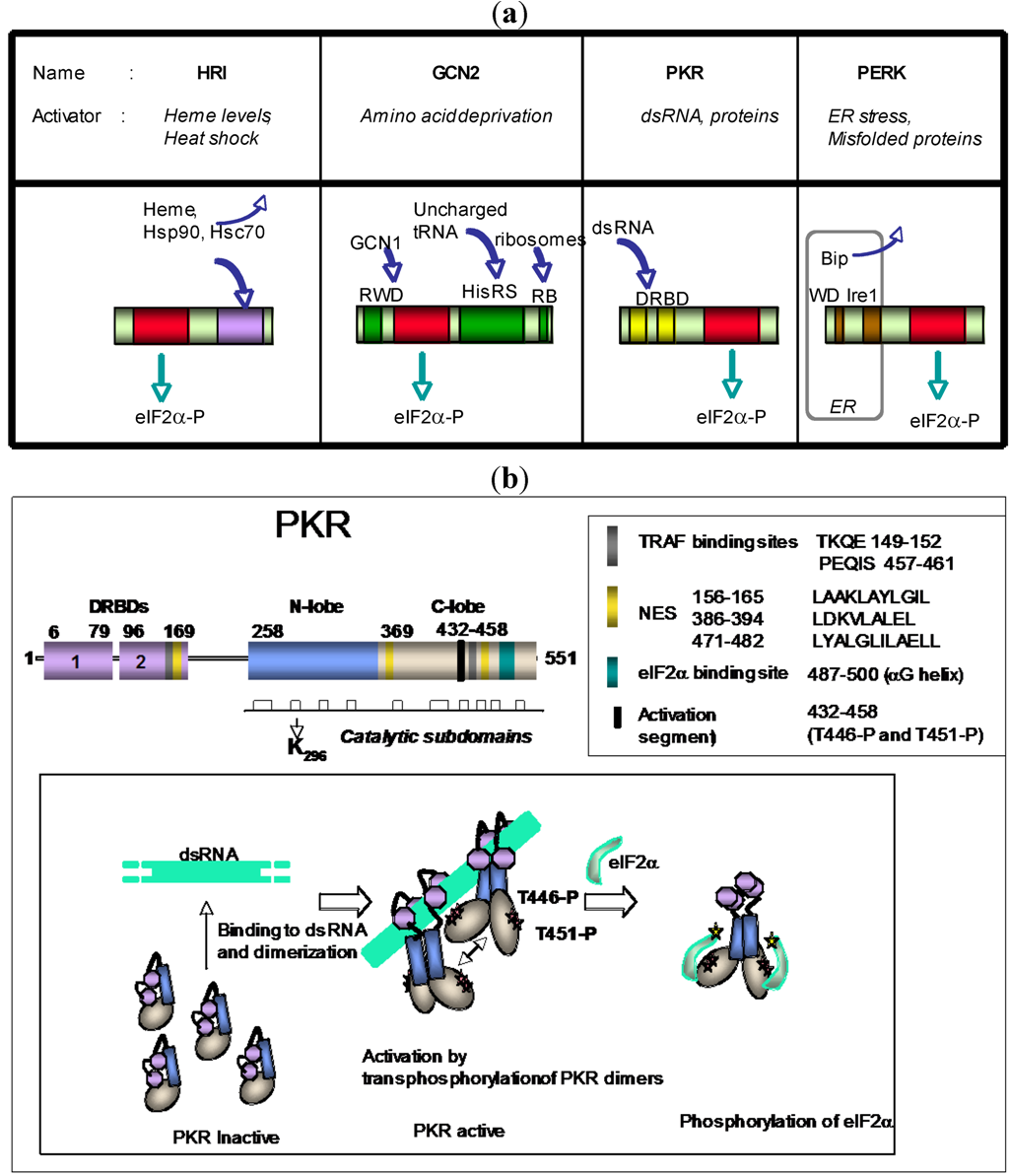
:1. Structure of PKR

2. Cellular Localization of PKR
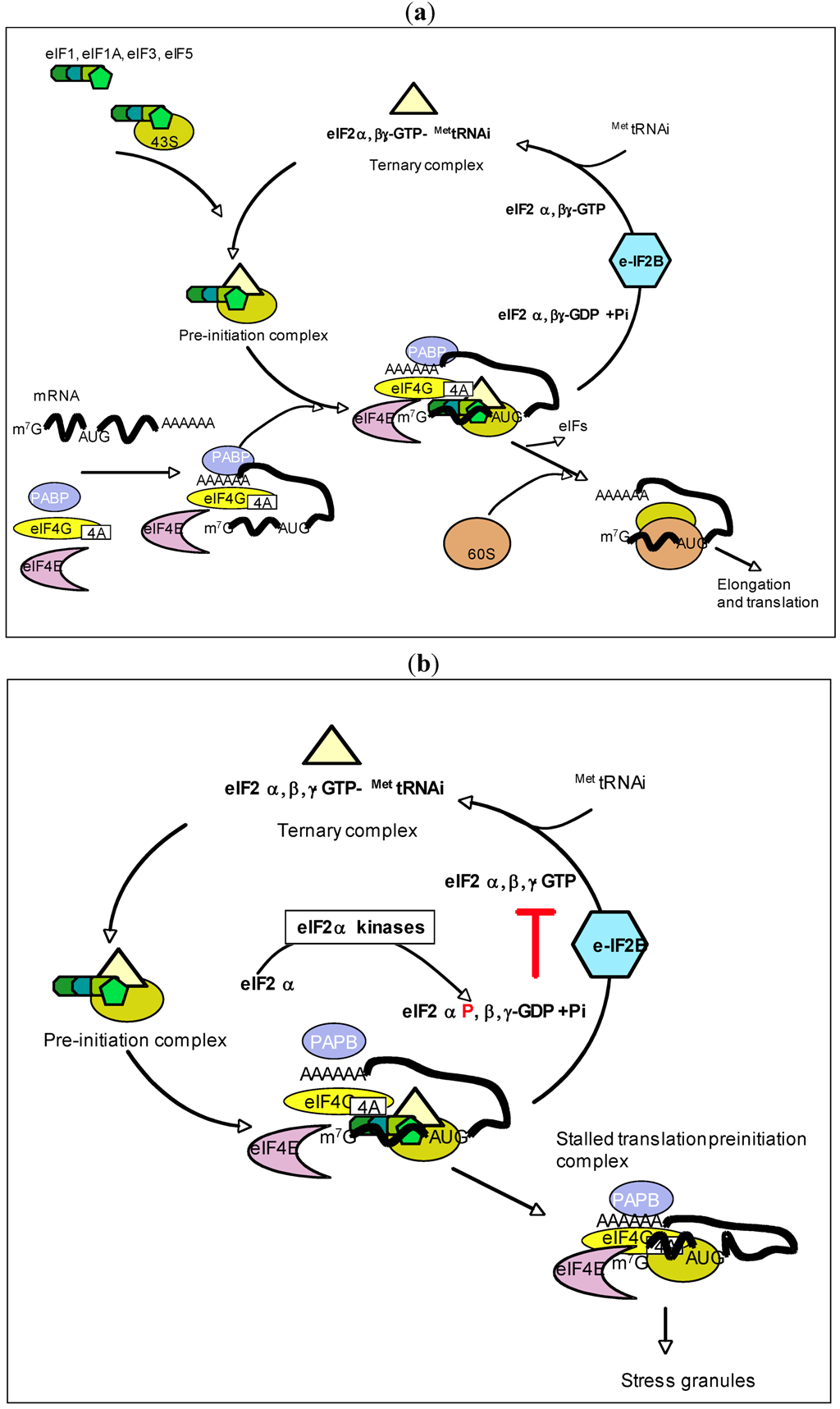
3. PKR and eIF2α-Kinase Activity
3.1. eIF2α-Kinase
3.2. eIF2α Phosphorylation and Viral Escape

3.3. eIF2α Phosphorylation and Impact on Cell Metabolism
3.4. eIF2α Phosphorylation and Autophagy
4. Physiological Role of PKR
4.2. PKR as a Cognitive Decline Biomarker
4.3. Regulation of Signaling Pathways
4.3.1. PKR and Tumor Suppressors
4.3.2. PKR and the NF-κB Pathway
4.3.3. PKR and MAPKinase Pathway
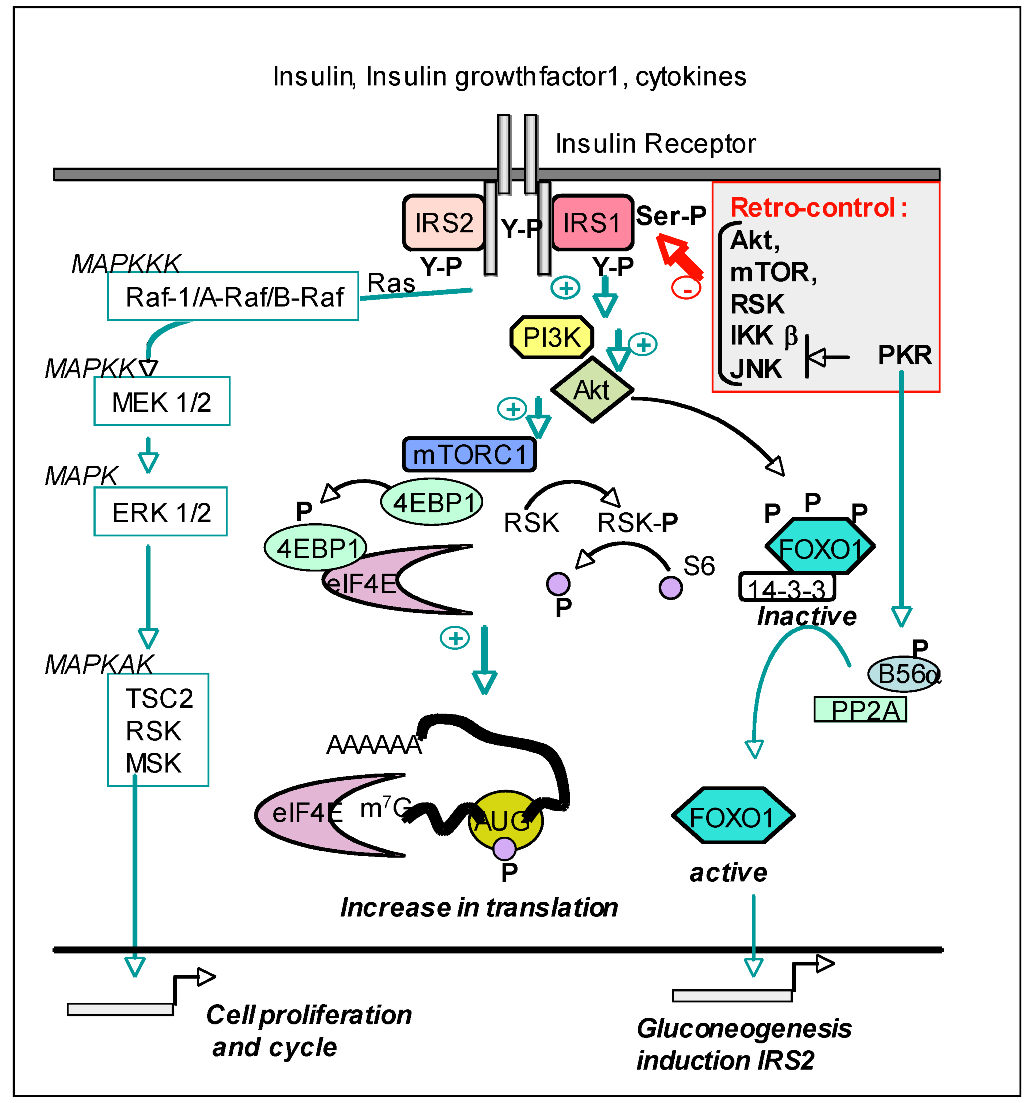
4.3.4. PKR and the Insulin Pathway

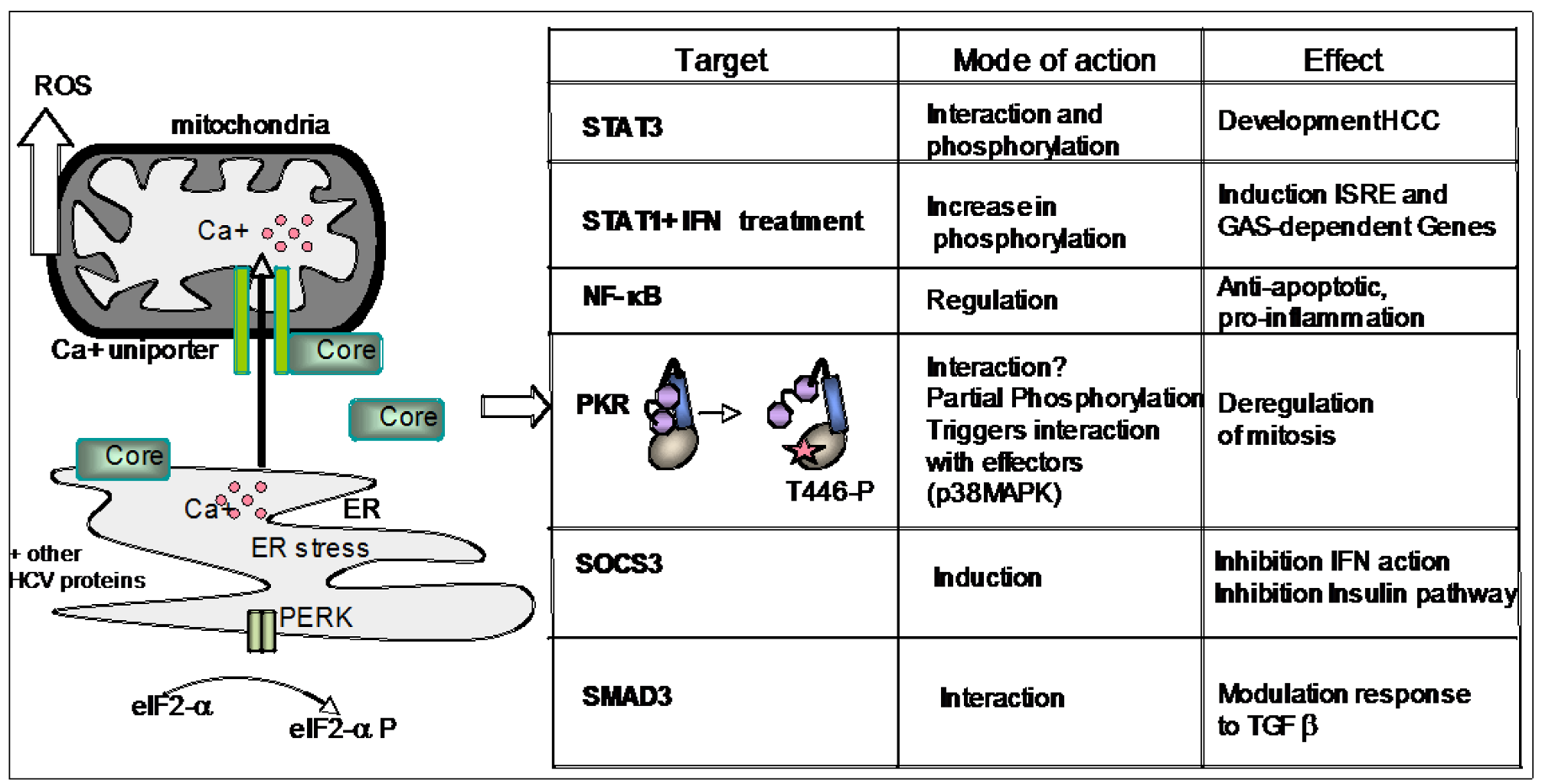
5. PKR and Its Role in HCV Infection
5.1. Interaction of HCV with PKR
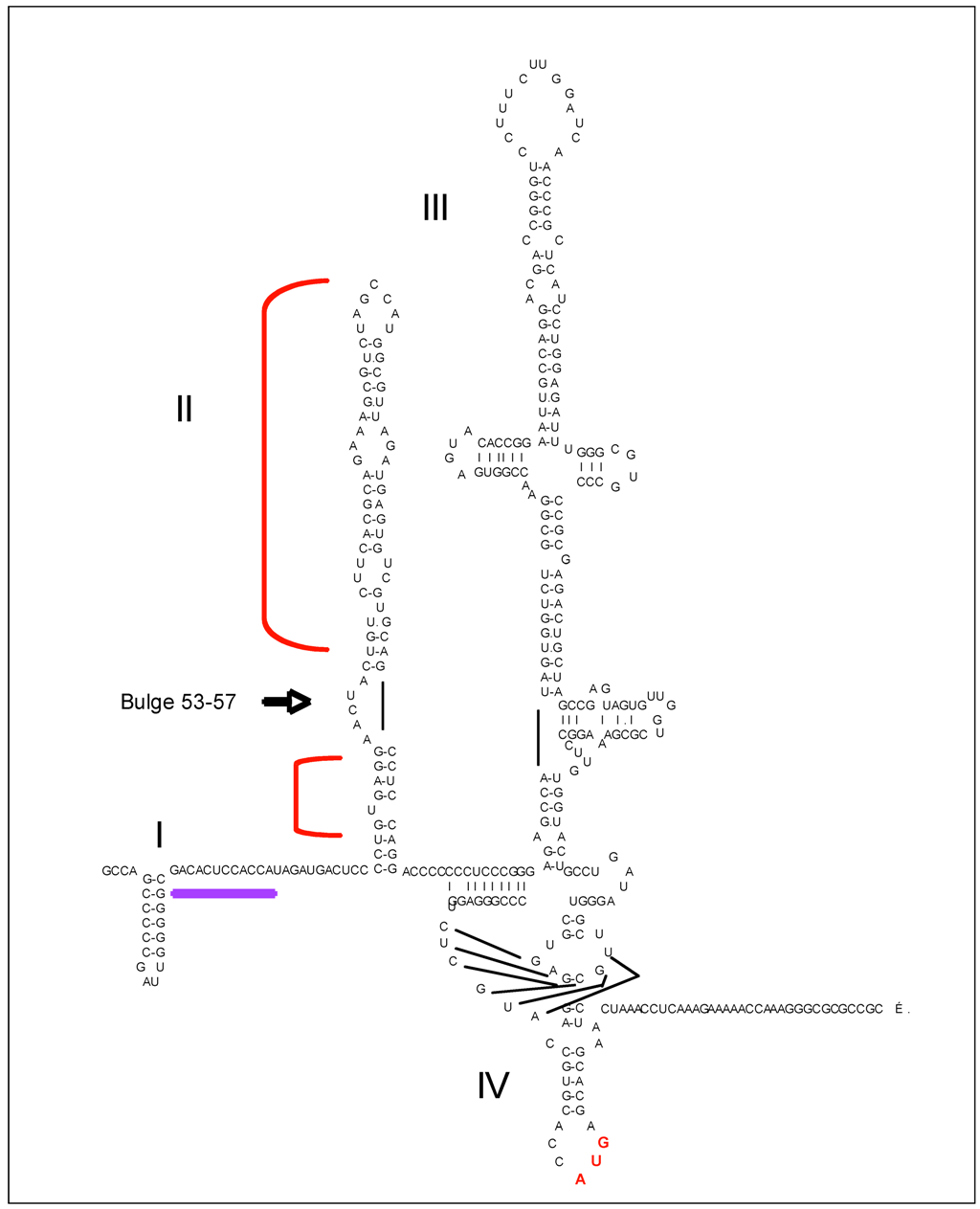
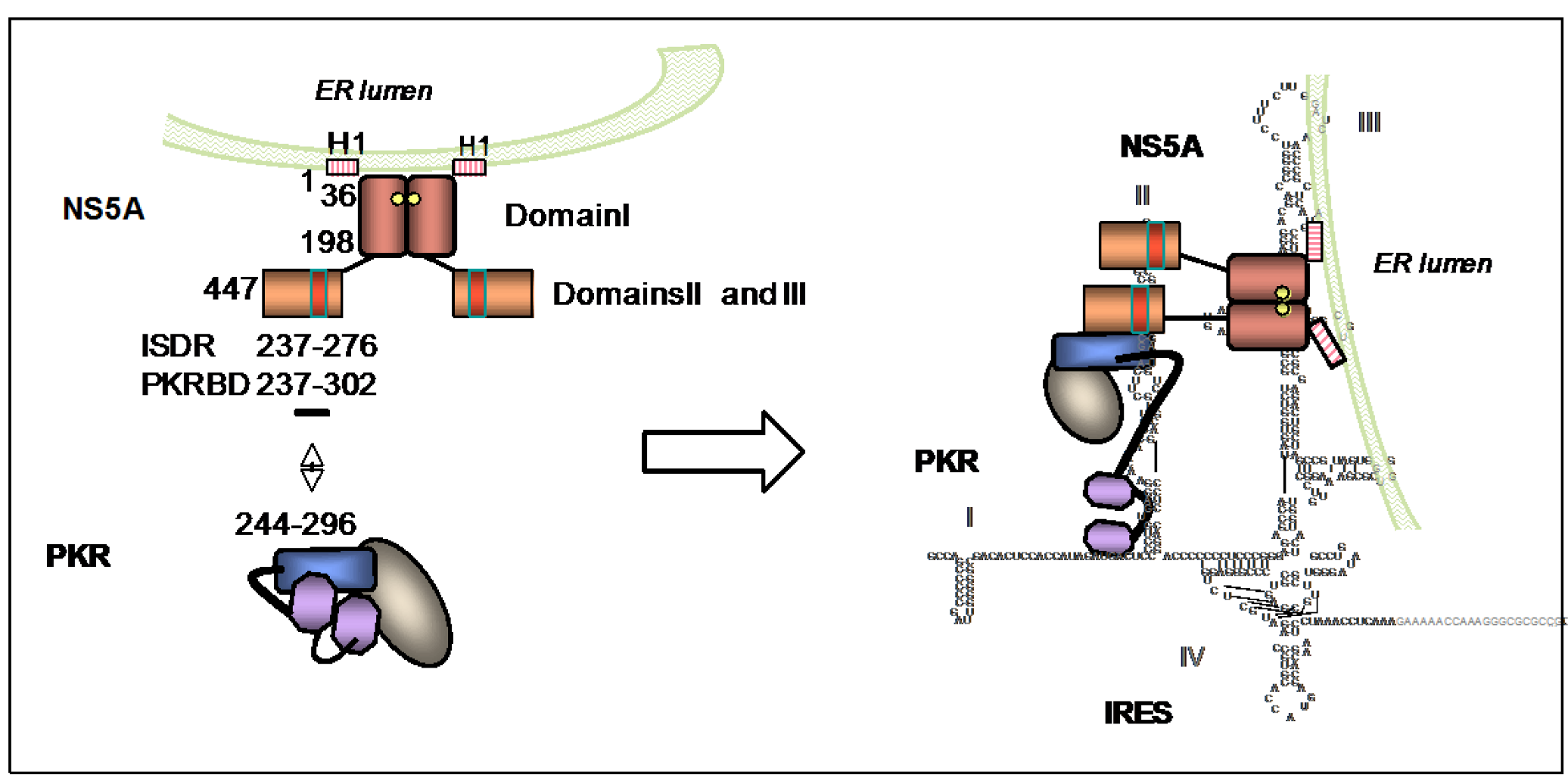
5.1.1. PKR and HCV IRES

5.1.2. PKR and Core Protein

5.1.3. PKR and NS5A

5.1.4. PKR and E2
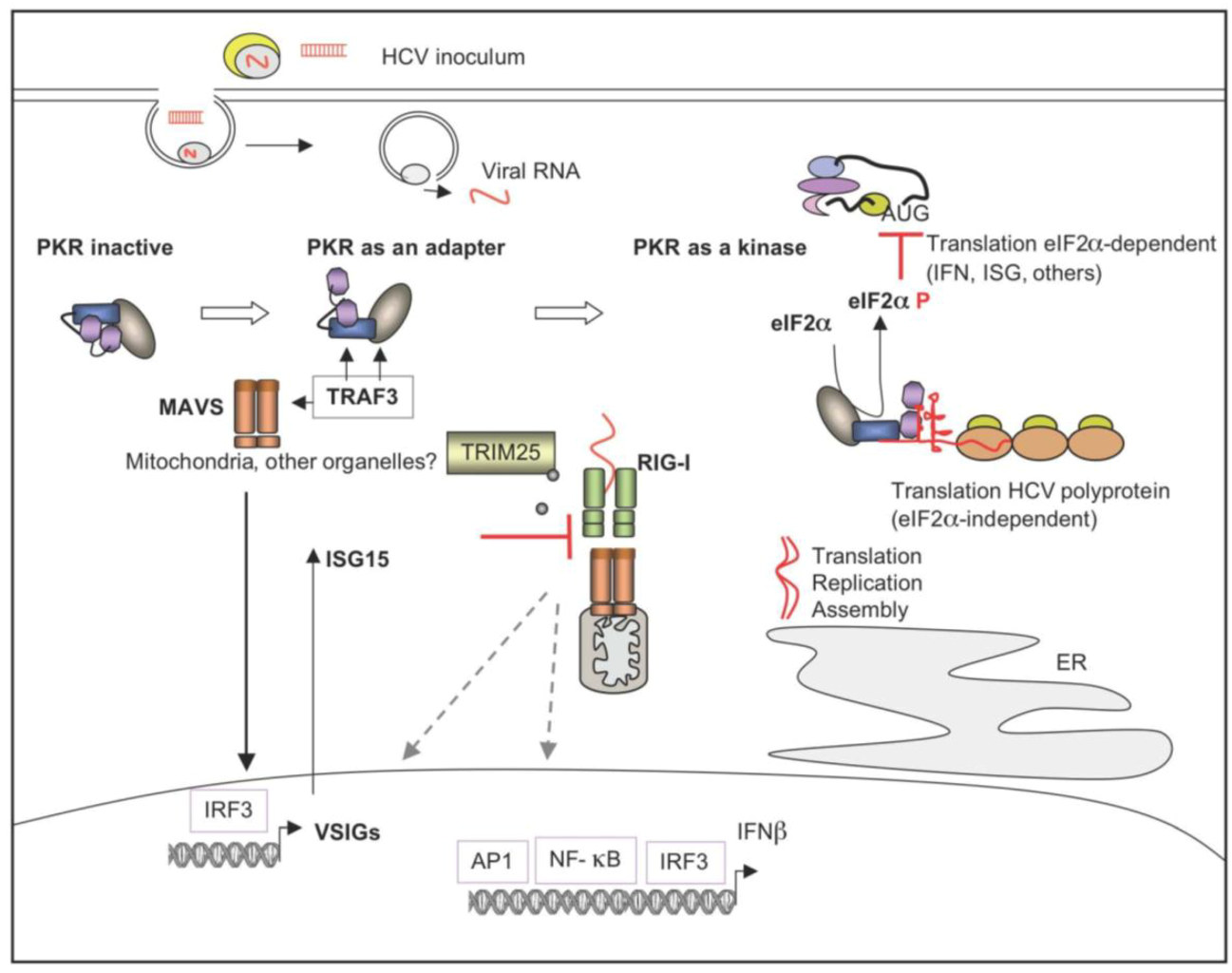
5.2. HCV and the Innate Immune Response
5.2.1. HCV and Activation of the Pathogen Recognition Receptors
5.2.2. Control of IFN Induction by HCV NS3/4A

5.2.3. HCV and Biomarkers Linked to the Immune Response
5.2.4. PKR and ISG15, Two ISGs as Pro-HCV Agents

5.2.5. PKR and Modulation of Pathways Related to Innate Immunity
5.2.6. A Role for Pharmalogical Inhibitors of PKR?
6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, S.B.; Kerr, I.M.; Williams, B.R.G.; Hovanessian, A.G. Molecular cloning and characterization of cDNA encoding human double-stranded RNA activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Raven, J.F.; Koromilas, A.E. PERK and PKR: Old kinases learn new tricks. Cell Cycle 2008, 7, 1146–1150. [Google Scholar] [CrossRef]
- Saunders, L.R.; Barber, G.N. The dsRNA binding protein family: Critical roles, diverse cellular functions. FASEB J. 2003, 17, 961–983. [Google Scholar]
- Daher, A.; Laraki, G.; Singh, M.; Melendez-Pena, C.E.; Bannwarth, S.; Peters, A.H.; Meurs, E.F.; Braun, R.E.; Patel, R.C.; Gatignol, A. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol. Cell Biol. 2009, 29, 254–265. [Google Scholar]
- Nallagatla, S.R.; Toroney, R.; Bevilacqua, P.C. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr. Opin. Struct. Biol. 2011, 21, 119–127. [Google Scholar] [CrossRef]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar]
- Katze, M.G.; Wambach, M.; Wong, M.L.; Garfinkel, M.; Meurs, E.; Chong, K.; Williams, B.R.G.; Hovanessian, A.G.; Barber, G.N. Functional expression and RNA binding analysis of the interferon-induced, dsRNA activated 68,000 Mr protein kinase in a cell free system. Mol. Cell Biol. 1991, 11, 5497–5505. [Google Scholar]
- Gil, J.; Garcia, M.A.; Gomez-Puertas, P.; Guerra, S.; Rullas, J.; Nakano, H.; Alcami, J.; Esteban, M. TRAF family proteins link PKR with NF-kappa B activation. Mol. Cell Biol. 2004, 24, 4502–4512. [Google Scholar] [CrossRef]
- Xu, Z.; Williams, B.R. Genomic features of human PKR: Alternative splicing and a polymorphic CGG repeat in the 5'-untranslated region. J. Interferon Cytokine Res. 1998, 18, 609–616. [Google Scholar] [CrossRef]
- Takizawa, T.; Tatematsu, C.; Watanabe, M.; Yoshida, M.; Nakajima, K. Three leucine-rich sequences and the N-terminal region of double-stranded RNA-activated protein kinase (PKR) are responsible for its cytoplasmic localization. J. Biochem. 2000, 128, 471–476. [Google Scholar] [CrossRef]
- la Cour, T.; Kiemer, L.; Molgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef]
- Pataer, A.; Vorburger, S.A.; Chada, S.; Balachandran, S.; Barber, G.N.; Roth, J.A.; Hunt, K.K.; Swisher, S.G. Melanoma differentiation-associated gene-7 protein physically associates with the double-stranded RNA-activated protein kinase PKR. Mol. Ther. 2005, 11, 717–723. [Google Scholar]
- Tian, B.; Mathews, M.B. Functional characterization of and cooperation between the double-stranded RNA-binding motifs of the protein kinase PKR. J. Biol. Chem. 2001, 276, 9936–9944. [Google Scholar]
- Bonnet, M.C.; Daurat, C.; Ottone, C.; Meurs, E.F. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal. 2006, 18, 1865–1875. [Google Scholar] [CrossRef]
- Wu, S.; Kumar, K.U.; Kaufman, R.J. Identification and requirement of three ribosome binding domains in dsRNA-dependent protein kinase (PKR). Biochemistry 1998, 37, 13816–13826. [Google Scholar]
- Besse, S.; Rebouillat, D.; Marie, I.; Puvion-Dutilleul, F.; Hovanessian, A.G. Ultrastructural localization of interferon-inducible double-stranded RNA-activated enzymes in human cells. Exp. Cell Res. 1998, 239, 379–392. [Google Scholar] [CrossRef]
- Jimenez-Garcia, L.F.; Green, S.R.; Mathews, M.B.; Spector, D.L. Organization of the double-stranded RNA-activated protein kinase DAI and virus-associated VA RNAI in adenovirus-2-infected HeLa cells. J. Cell Sci. 1993, 106, 11–22. [Google Scholar]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef]
- Jeffrey, I.W.; Kadereit, S.; Meurs, E.F.; Metzger, T.; Bachmann, M.; Schwemmle, M.; Hovanessian, A.G.; Clemens, M.J. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp. Cell Res. 1995, 218, 17–27. [Google Scholar] [CrossRef]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Martinelli, G.; Blalock, W.L.; Cocco, L.; Martelli, A.M. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia 2008, 22, 2267–2269. [Google Scholar]
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Tagliavini, F.; Faenza, I.; Martelli, A.M.; Follo, M.Y.; Cocco, L. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia 2011, 25, 236–245. [Google Scholar] [CrossRef]
- Bando, Y.; Onuki, R.; Katayama, T.; Manabe, T.; Kudo, T.; Taira, K.; Tohyama, M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson's disease and Huntington's disease. Neurochem. Int. 2005, 46, 11–18. [Google Scholar] [CrossRef]
- Bose, A.; Mouton-Liger, F.; Paquet, C.; Mazot, P.; Vigny, M.; Gray, F.; Hugon, J. Modulation of tau phosphorylation by the kinase PKR: Implications in Alzheimer's disease. Brain Pathol. 2010, 21, 189–200. [Google Scholar]
- Paquet, C.; Bose, A.; Polivka, M.; Peoc'h, K.; Brouland, J.P.; Keohane, C.; Hugon, J.; Gray, F. Neuronal phosphorylated RNA-dependent protein kinase in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 2009, 68, 190–198. [Google Scholar] [CrossRef]
- Farrell, P.J.; Balkow, K.; Hunt, T.; Jackson, R.J. Phosphorylation of initiation factor eIF2 and the control of reticulocyte protein synthesis. Cell 1977, 11, 187–200. [Google Scholar] [CrossRef]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Dey, M.; Trieselmann, B.; Locke, E.G.; Lu, J.; Cao, C.; Dar, A.C.; Krishnamoorthy, T.; Dong, J.; Sicheri, F.; Dever, T.E. PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2alpha. Mol. Cell Biol. 2005, 25, 3063–3075. [Google Scholar] [CrossRef]
- Chong, K.; Feng, L.; Donahue, T.F.; Friesen, J.D.; Meurs, E.; Hovanessian, A.G.; Williams, B.R.G. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992, 11, 1553–1562. [Google Scholar]
- Meurs, E.F.; Watanabe, Y.; Kadereit, S.; Barber, G.N.; Katze, M.G.; Chong, K.; Williams, B.R.G.; Hovanessian, A.G. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eucaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J. Virol. 1992, 66, 5805–5814. [Google Scholar]
- Dar, A.C.; Sicheri, F. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol. Cell 2002, 10, 295–305. [Google Scholar] [CrossRef]
- White, J.P.; Reineke, L.C.; Lloyd, R.E. Poliovirus switches to an eIF2-independent mode of translation during infection. J. Virol. 2011, 85, 8884–8893. [Google Scholar] [CrossRef]
- Wilson, J.E.; Pestova, T.V.; Hellen, C.U.; Sarnow, P. Initiation of protein synthesis from the A site of the ribosome. Cell 2000, 102, 511–520. [Google Scholar] [CrossRef]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef]
- Shimoike, T.; McKenna, S.A.; Lindhout, D.A.; Puglisi, J.D. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antivir. Res. 2009, 83, 228–237. [Google Scholar]
- Arnaud, N.; Dabo, S.; Maillard, P.; Budkowska, A.; Kalliampakou, K.I.; Mavromara, P.; Garcin, D.; Hugon, J.; Gatignol, A.; Akazawa, D.; et al. Hepatitis C virus controls interferon production through PKR activation. PLoS One 2010, 5, e10575. [Google Scholar]
- Kim, J.H.; Park, S.M.; Park, J.H.; Keum, S.J.; Jang, S.K. eIF2A mediates translation of hepatitis C viral mRNA under stress conditions. EMBO J. 2011, 30, 2454–2464. [Google Scholar]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Courtney, S.C.; Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. West nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 2012, 86, 3647–3657. [Google Scholar]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar]
- Yang, Y.L.; Reis, L.F.; Pavlovic, J.; Aguzzi, A.; Schafer, R.; Kumar, A.; Williams, B.R.; Aguet, M.; Weissmann, C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995, 14, 6095–6106. [Google Scholar]
- Ruggieri, A.; Dazert, E.; Metz, P.; Hofmann, S.; Bergeest, J.P.; Mazur, J.; Bankhead, P.; Hiet, M.S.; Kallis, S.; Alvisi, G.; et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 2012, 12, 71–85. [Google Scholar] [CrossRef]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 2012, 7, e43031. [Google Scholar]
- Hinnebusch, A.G. Evidence for translational regulation of the activator of general amino acid control in yeast. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 6442–6446. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- O'Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef]
- Paquet, C.; Mouton-Liger, F.; Meurs, E.F.; Mazot, P.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. The PKR activator PACT is induced by Abeta: Involvement in Alzheimer’s disease. Brain Pathol. 2011, 22, 219–229. [Google Scholar]
- Beau, I.; Mehrpour, M.; Codogno, P. Autophagosomes and human diseases. Int. J. Biochem. Cell Biol. 2011, 43, 460–464. [Google Scholar] [CrossRef]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.T.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 190–195. [Google Scholar]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef]
- Abraham, N.; Stojdl, D.F.; Duncan, P.I.; Methot, N.; Ishii, T.; Dube, M.; Vanderhyden, B.C.; Atkins, H.L.; Gray, D.A.; McBurney, M.W.; et al. Characterization of transgenic mice with targeted dsruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 1999, 274, 5953–5962. [Google Scholar]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar]
- Durbin, R.K.; Mertz, S.E.; Koromilas, A.E.; Durbin, J.E. PKR protection against intranasal vesicular stomatitis virus infection is mouse strain dependent. Viral Immunol. 2002, 15, 41–51. [Google Scholar] [CrossRef]
- Clerzius, G.; Gelinas, J.F.; Gatignol, A. Multiple levels of PKR inhibition during HIV-1 replication. Rev. Med. Virol. 2011, 21, 42–53. [Google Scholar] [CrossRef]
- Ong, C.L.; Thorpe, J.C.; Gorry, P.R.; Bannwarth, S.; Jaworowski, A.; Howard, J.L.; Chung, S.; Campbell, S.; Christensen, H.S.; Clerzius, G.; et al. Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J. Virol. 2005, 79, 12763–12772. [Google Scholar]
- Daniels, S.; Gatignol, A. The Multiple Functions of TRBP, at the Hub of Cell Responses to Viruses, Stress and Cancer. Microbiol. Mol. Biol. Rev. 2012, 76, 652–666. [Google Scholar] [CrossRef]
- Clerzius, G.; Gelinas, J.F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar]
- Nie, Y.; Hammond, G.L.; Yang, J.H. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 2007, 81, 917–923. [Google Scholar] [CrossRef]
- Li, Z.; Wolff, K.C.; Samuel, C.E. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 2010, 396, 316–322. [Google Scholar]
- Toth, A.M.; Li, Z.; Cattaneo, R.; Samuel, C.E. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 2009, 284, 29350–29356. [Google Scholar]
- Li, Z.; Okonski, K.M.; Samuel, C.E. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J. Virol. 2012, 86, 3787–3794. [Google Scholar]
- Gelinas, J.F.; Clerzius, G.; Shaw, E.; Gatignol, A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J. Virol. 2011, 85, 8460–8466. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012. [Google Scholar]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar]
- Zhu, P.J.; Huang, W.; Kalikulov, D.; Yoo, J.W.; Placzek, A.N.; Stoica, L.; Zhou, H.; Bell, J.C.; Friedlander, M.J.; Krnjevic, K.; et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-gamma-mediated disinhibition. Cell 2011, 147, 1384–1396. [Google Scholar] [CrossRef]
- Damjanac, M.; Page, G.; Ragot, S.; Laborie, G.; Gil, R.; Hugon, J.; Paccalin, M. PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J. Cell. Mol. Med. 2009, 13, 1823–1832. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Roy, S.; Barber, G.N.; Katze, M.G.; Sonenberg, N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science 1992, 257, 1685–1689. [Google Scholar]
- Meurs, E.F.; Galabru, J.; Barber, G.N.; Katze, M.G.; Hovanessian, A.G. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 232–236. [Google Scholar]
- Cuddihy, A.R.; Wong, A.H.; Tam, N.W.; Li, S.; Koromilas, A.E. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 1999, 18, 2690–2702. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Li, S.; Tam, N.W.; Wong, A.H.; Taya, Y.; Abraham, N.; Bell, J.C.; Koromilas, A.E. Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol. Cell Biol. 1999, 19, 2475–2484. [Google Scholar]
- Bennett, R.L.; Pan, Y.; Christian, J.; Hui, T.; May, W.S., Jr. The RAX/PACT-PKR stress response pathway promotes p53 sumoylation and activation, leading to G(1) arrest. Cell Cycle 2012, 11, 407–417. [Google Scholar] [CrossRef]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 6288–6292. [Google Scholar]
- Kumar, A.; Yang, Y.L.; Flati, V.; Der, S.; Kadereit, S.; Deb, A.; Haque, J.; Reis, L.; Weissmann, C.; Williams, B.R. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: Role of IRF-1 and NF-kappaB. EMBO J. 1997, 16, 406–416. [Google Scholar]
- Zamanian-daryoush, M.; Mogensen, T.H.; Didonato, J.A.; Williams, B.R.G. NF-kB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kB-inducing kinase and IkB kinase. Mol. Cell Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef]
- Chu, W.M.; Ostertag, D.; Li, Z.W.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef]
- Bonnet, M.C.; Weil, R.; Dam, E.; Hovanessian, A.G.H.; Meurs, E.F. PKR stimulates NF-kB irrespective of its kinase function by interacting with the IkB kinase complex. Mol. Cell Biol. 2000, 20, 4532–4542. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Iordanov, M.S.; Paranjape, J.M.; Zhou, A.; Wong, J.; Williams, B.R.G.; Meurs, E.F.; SIlverman, R.H.; Magun, B.E. Activation of p38 mitogen-activated protein kinase and c-jun NH2-terminal kinase by double-stranded RNA and encephalomyocarditis virus: Involvement of RNAse L, protein kinase R and alternative pathways. Mol. Cell Biol. 2000, 20, 617–627. [Google Scholar] [CrossRef]
- Yoshida, R.; Takaesu, G.; Yoshida, H.; Okamoto, F.; Yoshioka, T.; Choi, Y.; Akira, S.; Kawai, T.; Yoshimura, A.; Kobayashi, T. TRAF6 and MEKK1 play a pivotal role in the RIG-I-like helicase antiviral pathway. J. Biol. Chem. 2008, 283, 36211–36220. [Google Scholar]
- Mikkelsen, S.S.; Jensen, S.B.; Chiliveru, S.; Melchjorsen, J.; Julkunen, I.; Gaestel, M.; Arthur, J.S.; Flavell, R.A.; Ghosh, S.; Paludan, S.R. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: Dependence on TRAF2 and TAK1. J. Biol. Chem. 2009, 284, 10774–10782. [Google Scholar]
- Silva, A.M.; Whitmore, M.; Xu, Z.; Jiang, Z.; Li, X.; Williams, B.R. Protein kinase R (PKR) interacts with and activates mitogen-activated protein kinase kinase 6 (MKK6) in response to double-stranded RNA stimulation. J. Biol. Chem. 2004, 279, 37670–37676. [Google Scholar]
- Zhang, P.; Langland, J.O.; Jacobs, B.L.; Samuel, C.E. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J. Virol. 2009, 83, 5718–5725. [Google Scholar] [CrossRef]
- Myskiw, C.; Arsenio, J.; van Bruggen, R.; Deschambault, Y.; Cao, J. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathway. J. Virol. 2009, 83, 6757–6768. [Google Scholar]
- Takada, Y.; Ichikawa, H.; Pataer, A.; Swisher, S.; Aggarwal, B.B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene 2007, 26, 1201–1212. [Google Scholar] [CrossRef]
- Corradetti, M.N.; Guan, K.L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef]
- Yang, X.; Nath, A.; Opperman, M.J.; Chan, C. The double-stranded RNA-dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells. Mol. Biol. Cell 2010, 21, 3449–3458. [Google Scholar]
- Chang, J.; Guo, J.T.; Jiang, D.; Guo, H.; Taylor, J.M.; Block, T.M. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J. Virol. 2008, 82, 8215–8223. [Google Scholar] [CrossRef]
- Toroney, R.; Nallagatla, S.R.; Boyer, J.A.; Cameron, C.E.; Bevilacqua, P.C. Regulation of PKR by HCV IRES RNA: Importance of domain II and NS5A. J. Mol. Biol. 2010, 400, 393–412. [Google Scholar] [CrossRef]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 1992, 12, 5238–5248. [Google Scholar]
- Wang, C.; Pflugheber, J.; Sumpter, R., Jr.; Sodora, D.L.; Hui, D.; Sen, G.C.; Gale, M., Jr. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J. Virol. 2003, 77, 3898–3912. [Google Scholar] [CrossRef]
- Taylor, D.R.; Puig, M.; Darnell, M.E.; Mihalik, K.; Feinstone, S.M. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005, 79, 6291–6298. [Google Scholar]
- Kang, J.I.; Kwon, S.N.; Park, S.H.; Kim, Y.K.; Choi, S.Y.; Kim, J.P.; Ahn, B.Y. PKR protein kinase is activated by hepatitis C virus and inhibits viral replication through translational control. Virus Res. 2009, 142, 51–56. [Google Scholar] [CrossRef]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef]
- Arnaud, N.; Dabo, S.; Akazawa, D.; Fukasawa, M.; Shinkai-Ouchi, F.; Hugon, J.; Wakita, T.; Meurs, E.F. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011, 7, e1002289. [Google Scholar] [CrossRef]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- McLauchlan, J. Hepatitis C virus: viral proteins on the move. Biochem. Soc. Trans. 2009, 37, 986–990. [Google Scholar] [CrossRef]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar]
- Yoshida, H.; Kato, N.; Shiratori, Y.; Otsuka, M.; Maeda, S.; Kato, J.; Omata, M. Hepatitis C virus core protein activates nuclear factor kappa B-dependent signaling through tumor necrosis factor receptor-associated factor. J. Biol. Chem. 2001, 276, 16399–16405. [Google Scholar]
- Joo, M.; Hahn, Y.S.; Kwon, M.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. J. Virol. 2005, 79, 7648–7657. [Google Scholar]
- Basu, A.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein modulates the interferon-induced transacting factors of Jak/Stat signaling pathway but does not affect the activation of downstream IRF-1 or 561 gene. Virology 2001, 288, 379–390. [Google Scholar]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef]
- Yoshida, T.; Hanada, T.; Tokuhisa, T.; Kosai, K.; Sata, M.; Kohara, M.; Yoshimura, A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 2002, 196, 641–653. [Google Scholar] [CrossRef]
- Larrea, E.; Aldabe, R.; Molano, E.; Fernandez-Rodriguez, C.M.; Ametzazurra, A.; Civeira, M.P.; Prieto, J. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: in vivo and in vitro studies. Gut 2006, 55, 1188–1196. [Google Scholar]
- Bode, J.G.; Ludwig, S.; Ehrhardt, C.; Albrecht, U.; Erhardt, A.; Schaper, F.; Heinrich, P.C.; Haussinger, D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003, 17, 488–490. [Google Scholar]
- Kawaguchi, T.; Yoshida, T.; Harada, M.; Hisamoto, T.; Nagao, Y.; Ide, T.; Taniguchi, E.; Kumemura, H.; Hanada, S.; Maeyama, M.; et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 2004, 165, 1499–1508. [Google Scholar] [CrossRef]
- Cheng, P.L.; Chang, M.H.; Chao, C.H.; Lee, Y.H. Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF-beta/Smad3-mediated transcriptional activation. Oncogene 2004, 23, 7821–7838. [Google Scholar] [CrossRef]
- Battaglia, S.; Benzoubir, N.; Nobilet, S.; Charneau, P.; Samuel, D.; Zignego, A.L.; Atfi, A.; Brechot, C.; Bourgeade, M.F. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS One 2009, 4, e4355. [Google Scholar]
- Alisi, A.; Mele, R.; Spaziani, A.; Tavolaro, S.; Palescandolo, E.; Balsano, C. Thr 446 phosphorylation of PKR by HCV core protein deregulates G2/M phase in HCC cells. J. Cell. Physiol. 2005, 205, 25–31. [Google Scholar]
- Spaziani, A.; Alisi, A.; Sanna, D.; Balsano, C. Role of p38 MAPK and RNA-dependent protein kinase (PKR) in hepatitis C virus core-dependent nuclear delocalization of cyclin B1. J. Biol. Chem. 2006, 281, 10983–10989. [Google Scholar] [CrossRef]
- Yang, X.B.; Battaglia, S.; Boucreux, D.; Chen, Z.; Brechot, C.; Pavio, N. Mapping of the interacting domains of hepatitis C virus core protein and the double-stranded RNA-activated protein kinase PKR. Virus Res. 2007, 125, 79–87. [Google Scholar] [CrossRef]
- Moriishi, K.; Okabayashi, T.; Nakai, K.; Moriya, K.; Koike, K.; Murata, S.; Chiba, T.; Tanaka, K.; Suzuki, R.; Suzuki, T.; et al. Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J. Virol. 2003, 77, 10237–10249. [Google Scholar]
- Cerutti, A.; Maillard, P.; Minisini, R.; Vidalain, P.O.; Roohvand, F.; Pecheur, E.I.; Pirisi, M.; Budkowska, A. Identification of a functional, CRM-1-dependent nuclear export signal in hepatitis C virus core protein. PLoS One 2011, 6, e25854. [Google Scholar]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakani, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5A gene response to interferon in patients with chronic Hepatitis C virus 1b infection. New Engl. J. Med. 1996, 334, 77–81. [Google Scholar]
- Duverlie, G.; Khorsi, H.; Castelain, S.; Jaillon, O.; Izopet, J.; Lunel, F.; Eb, F.; Penin, F.; Wychowski, C. Sequence analysis of the NS5A protein of European hepatitis C virus 1b isolates and relation to interferon sensitivity. J. Gen. Virol. 1998, 79, 1373–1381. [Google Scholar]
- Zeuzem, S.; Lee, J.H.; Roth, W.K. Mutations in the non structural 5A gene of european hepatits C virus isolates and response to interferon alfa. Hepatology 1997, 25, 740–749. [Google Scholar]
- Donlin, M.J.; Cannon, N.A.; Aurora, R.; Li, J.; Wahed, A.S.; Di Bisceglie, A.M.; Tavis, J.E. Contribution of genome-wide HCV genetic differences to outcome of interferon-based therapy in Caucasian American and African American patients. PLoS One 2010, 5, e9032. [Google Scholar]
- Gale, M.J., Jr.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Blakely, C.M.; Kwieciszewski, B.; Tan, S.L.; Dossett, M.; Tang, N.M.; Korth, M.J.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: Molecular mechanisms of kinase regulation. Mol. Cell. Biol. 1998, 18, 5208–5218. [Google Scholar]
- François, C.; Duverlie, G.; Rebouillat, D.; Khorsi, H.; Castelain, S.; Blum, H.E.; Gatignol, A.; Wychowski, C.; Moradpour, D.; Meurs, E.F. Expression of hepatitis C virus proteins interferes with the antiviral action of interferon independently of PKR-mediated control of protein synthesis. J. Virol. 2000, 74, 5587–5596. [Google Scholar]
- He, Y.; Tan, S.L.; Tareen, S.U.; Vijaysri, S.; Langland, J.O.; Jacobs, B.L.; Katze, M.G. Regulation of mRNA translation and cellular signaling by hepatitis C virus nonstructural protein NS5A. J. Virol. 2001, 75, 5090–5098. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, W.C.; Huang, J.Y.; Chao, T.C.; Jeng, K.S.; Machida, K.; Lai, M.M. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J. Virol. 2010, 84, 7983–7993. [Google Scholar] [CrossRef]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar]
- Liang, Y.; Ye, H.; Kang, C.B.; Yoon, H.S. Domain 2 of nonstructural protein 5A (NS5A) of hepatitis C virus is natively unfolded. Biochemistry 2007, 46, 11550–11558. [Google Scholar] [CrossRef]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar]
- Taylor, D.R.; Tian, B.; Romano, P.R.; Hinnebusch, A.G.; Lai, M.M.; Mathews, M.B. Hepatitis C virus envelope protein E2 does not inhibit PKR by simple competition with autophosphorylation sites in the RNA-binding domain. J. Virol. 2001, 75, 1265–1273. [Google Scholar]
- Pavio, N.; Taylor, D.R.; Lai, M.M.C. Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J. Virol. 2002, 76, 1265–1272. [Google Scholar]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus Envelope protein E2 through the Eukaryotic Initiation factor 2a kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar]
- Bolcic, F.; Laufer, N.; Torres, C.; Cassino, L.; Reynoso, R.; Quarleri, J. Longitudinal analysis of the 5' UTR, E2-PePHD and NS5A-PKRBD genomic regions of hepatitis C virus genotype 1a in association with the response to peginterferon and ribavirin therapy in HIV-coinfected patients. Antivir. Res. 2012, 95, 72–81. [Google Scholar]
- Pang, P.S.; Planet, P.J.; Glenn, J.S. The evolution of the major hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS One 2009, 4, e6579. [Google Scholar] [CrossRef]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Matsumoto, M.; Oshiumi, H.; Seya, T. Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. 2011, 21, 67–77. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef]
- Binder, M.; Kochs, G.; Bartenschlager, R.; Lohmann, V. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 2007, 46, 1365–1374. [Google Scholar]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef]
- Uzri, D.; Gehrke, L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J. Virol. 2009, 83, 4174–4184. [Google Scholar] [CrossRef]
- Binder, M.; Eberle, F.; Seitz, S.; Mucke, N.; Huber, C.M.; Kiani, N.; Kaderali, L.; Lohmann, V.; Dalpke, A.; Bartenschlager, R. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 286, 27278–27287.
- Wang, N.; Liang, Y.; Devaraj, S.; Wang, J.; Lemon, S.M.; Li, K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 2009, 83, 9824–9834. [Google Scholar]
- Ebihara, T.; Shingai, M.; Matsumoto, M.; Wakita, T.; Seya, T. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology 2008, 48, 48–58. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Guo, Y.J.; Bin, L.; Sun, S.H. Hepatitis C virus single-stranded RNA induces innate immunity via Toll-like receptor 7. J. Hepatol. 2009, 51, 29–38. [Google Scholar] [CrossRef]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7431–7436. [Google Scholar]
- Ferreon, J.C.; Ferreon, A.C.; Li, K.; Lemon, S.M. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J. Biol. Chem. 2005, 280, 20483–20492. [Google Scholar]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2992–2997. [Google Scholar]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Lin, R.; Lacoste, J.; Nakhaei, P.; Sun, Q.; Yang, L.; Paz, S.; Wilkinson, P.; Julkunen, I.; Vitour, D.; Meurs, E.; et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3–4A proteolytic cleavage. J. Virol. 2006, 80, 6072–6083. [Google Scholar] [CrossRef]
- Bellecave, P.; Sarasin-Filipowicz, M.; Donze, O.; Kennel, A.; Gouttenoire, J.; Meylan, E.; Terracciano, L.; Tschopp, J.; Sarrazin, C.; Berg, T.; et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 2010, 51, 1127–1136. [Google Scholar]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell 2005, 122, 1–14. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.-Y.; Han, K.-J.; Lii, L.-Y.; Zhai, Z.; Shu, H.-B. VISA is an adapter protein required for Virus-Triggered IFN-b signaling. Mol. Cell 2005, 19, 1–14. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar]
- Mihm, S.; Frese, M.; Meier, V.; Wietzke-Braun, P.; Scharf, J.G.; Bartenschlager, R.; Ramadori, G. Interferon type I gene expression in chronic hepatitis C. Lab. Invest. 2004, 84, 1148–1159. [Google Scholar]
- Sarasin-Filipowicz, M.; Oakeley, E.J.; Duong, F.H.; Christen, V.; Terracciano, L.; Filipowicz, W.; Heim, M.H. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7034–7039. [Google Scholar]
- Bigger, C.B.; Guerra, B.; Brasky, K.M.; Hubbard, G.; Beard, M.R.; Luxon, B.A.; Lemon, S.M.; Lanford, R.E. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 2004, 78, 13779–13792. [Google Scholar]
- Patzwahl, R.; Meier, V.; Ramadori, G.; Mihm, S. Enhanced expression of interferon-regulated genes in the liver of patients with chronic hepatitis C virus infection: detection by suppression-subtractive hybridization. J. Virol. 2001, 75, 1332–1338. [Google Scholar] [CrossRef]
- Lagging, M.; Romero, A.I.; Westin, J.; Norkrans, G.; Dhillon, A.P.; Pawlotsky, J.M.; Zeuzem, S.; von Wagner, M.; Negro, F.; Schalm, S.W.; et al. IP-10 predicts viral response and therapeutic outcome in difficult-to-treat patients with HCV genotype 1 infection. Hepatology 2006, 44, 1617–1625. [Google Scholar]
- Casrouge, A.; Decalf, J.; Ahloulay, M.; Lababidi, C.; Mansour, H.; Vallet-Pichard, A.; Mallet, V.; Mottez, E.; Mapes, J.; Fontanet, A.; et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J. Clin. Invest. 2011, 121, 308–317. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar]
- Sarasin-Filipowicz, M.; Wang, X.; Yan, M.; Duong, F.H.; Poli, V.; Hilton, D.J.; Zhang, D.E.; Heim, M.H. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell Biol. 2009, 29, 4841–4851. [Google Scholar]
- Makowska, Z.; Duong, F.H.; Trincucci, G.; Tough, D.F.; Heim, M.H. Interferon-beta and interferon-lambda signaling is not affected by interferon-induced refractoriness to interferon-alpha in vivo. Hepatology 2011, 53, 1154–1163. [Google Scholar]
- Francois-Newton, V.; Magno de Freitas Almeida, G.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uze, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One 2011, 6, e22200. [Google Scholar]
- Muir, A.J.; Shiffman, M.L.; Zaman, A.; Yoffe, B.; de la Torre, A.; Flamm, S.; Gordon, S.C.; Marotta, P.; Vierling, J.M.; Lopez-Talavera, J.C.; et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 2010, 52, 822–832. [Google Scholar]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef]
- Payer, B.A.; Reiberger, T.; Aberle, J.; Ferenci, P.; Holzmann, H.; Rieger, A.; Peck-Radosavljevic, M. IL28B and interferon-gamma inducible protein 10 for prediction of rapid virologic response and sustained virologic response in HIV-HCV-coinfected patients. Eur. J. Clin. Invest. 2012, 42, 599–606. [Google Scholar] [CrossRef]
- Terenin, I.M.; Dmitriev, S.E.; Andreev, D.E.; Shatsky, I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 2008, 15, 836–841. [Google Scholar]
- Balvay, L.; Soto Rifo, R.; Ricci, E.P.; Decimo, D.; Ohlmann, T. Structural and functional diversity of viral IRESes. Biochim. Biophys. Acta 2009, 1789, 542–557. [Google Scholar] [CrossRef]
- Akazawa, D.; Date, T.; Morikawa, K.; Murayama, A.; Miyamoto, M.; Kaga, M.; Barth, H.; Baumert, T.F.; Dubuisson, J.; Wakita, T. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 2007, 81, 5036–5045. [Google Scholar] [CrossRef]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar]
- Farell, P.J.; Broeze, R.J.; Lengyel, P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature (London) 1979, 279, 523–524. [Google Scholar]
- Haas, A.L.; Ahrens, P.; Bright, P.M.; Ankel, H. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem. 1987, 262, 11315–11323. [Google Scholar]
- Elco, C.P.; Guenther, J.M.; Williams, B.R.G.; Sen, G.C. Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-kappaB, and interferon but not toll-like receptor 3. J. Virol. 2005, 79, 3920–3929. [Google Scholar] [CrossRef]
- Chen, L.; Sun, J.; Meng, L.; Heathcote, J.; Edwards, A.; McGilvray, I. ISG15, a ubiquitin-like interferon stimulated gene, promotes Hepatitis C Virus production in vitro: Implications for chronic infection and response to treatment. J. Gen. Virol. 2010, 91, 382–388. [Google Scholar] [CrossRef]
- Broering, R.; Zhang, X.; Kottilil, S.; Trippler, M.; Jiang, M.; Lu, M.; Gerken, G.; Schlaak, J.F. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the IFN response. Gut 2010, 59, 1111–1119. [Google Scholar] [CrossRef]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar]
- Zou, W.; Wang, J.; Zhang, D.E. Negative regulation of ISG15 E3 ligase EFP through its autoISGylation. Biochem. Biophys. Res. Commun. 2007, 354, 321–327. [Google Scholar] [CrossRef]
- Ruscanu, S.; Pascale, F.; Bourge, M.; Hemati, B.; Elhmouzi-Younes, J.; Urien, C.; Bonneau, M.; Takamatsu, H.; Hope, J.; Mertens, P.; et al. The double-stranded RNA bluetongue virus induces type I interferon in plasmacytoid dendritic cells via a MYD88-dependent TLR7/8-independent signaling pathway. J. Virol. 2012, 86, 5817–5828. [Google Scholar] [CrossRef]
- Irving, A.T.; Wang, D.; Vasilevski, O.; Latchoumanin, O.; Kozer, N.; Clayton, A.H.; Szczepny, A.; Morimoto, H.; Xu, D.; Williams, B.R.; et al. Regulation of actin dynamics by protein kinase R control of gelsolin enforces Basal innate immune defense. Immunity 2012, 36, 795–806. [Google Scholar] [CrossRef]
- Hugon, J.; Paquet, C.; Chang, R.C. Could PKR inhibition modulate human neurodegeneration? Expert. Rev. Neurother. 2009, 9, 1455–1457. [Google Scholar] [CrossRef]
- Nanduri, S.; Carpick, B.W.; Yang, Y.; Williams, B.R.G.; Qin, J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998, 17, 5458–5465. [Google Scholar] [CrossRef]
- Nekhai, S.; Bottaro, D.P.; Woldehawariat, G.; Spellerberg, A.; Petryshyn, R. A cell-permeable peptide inhibits activation of PKR and enhances cell proliferation. Peptides 2000, 21, 1449–1456. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dabo, S.; Meurs, E.F. dsRNA-Dependent Protein Kinase PKR and its Role in Stress, Signaling and HCV Infection. Viruses 2012, 4, 2598-2635. https://doi.org/10.3390/v4112598
Dabo S, Meurs EF. dsRNA-Dependent Protein Kinase PKR and its Role in Stress, Signaling and HCV Infection. Viruses. 2012; 4(11):2598-2635. https://doi.org/10.3390/v4112598
Chicago/Turabian StyleDabo, Stéphanie, and Eliane F. Meurs. 2012. "dsRNA-Dependent Protein Kinase PKR and its Role in Stress, Signaling and HCV Infection" Viruses 4, no. 11: 2598-2635. https://doi.org/10.3390/v4112598
APA StyleDabo, S., & Meurs, E. F. (2012). dsRNA-Dependent Protein Kinase PKR and its Role in Stress, Signaling and HCV Infection. Viruses, 4(11), 2598-2635. https://doi.org/10.3390/v4112598