HIV-1 Induced Bystander Apoptosis

{kind=link}
Abstract
:1. Introduction
2. Bystander Apoptosis
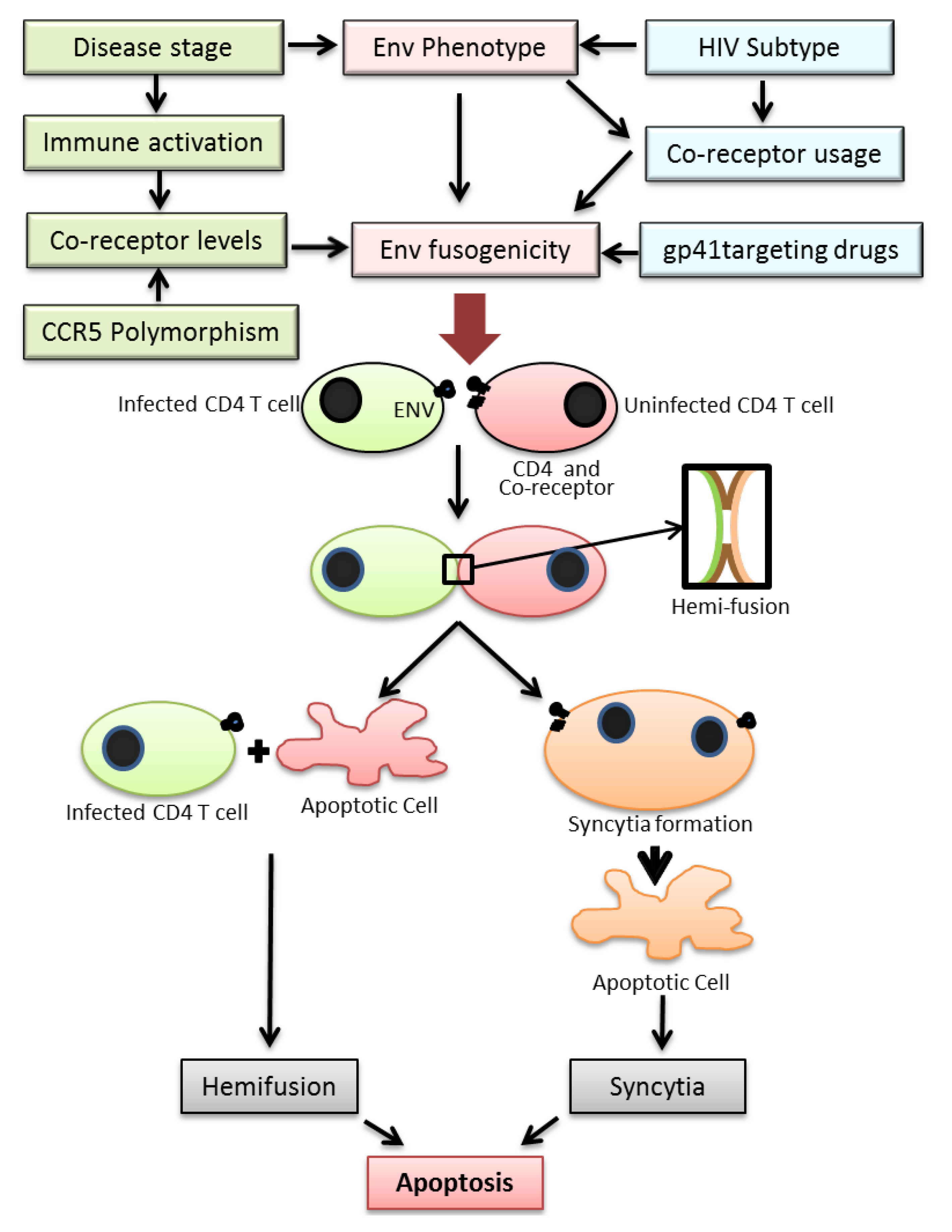
3. Env Glycoprotein Mediated Fusion
4. Env Glycoprotein Mediated Bystander Apoptosis
4.1. Apoptosis of Syncytia Formed via gp41 Mediated Fusion

4.2. Hemifusion Induced Apoptosis
5. Env Glycoprotein Phenotype and Bystander Apoptosis
5.1. Env Fusogenicity
5.2. Co-Receptor Usage
5.3. Disease Stage
5.4. HIV Subtype
6. Co-Receptor Expression Levels and Bystander Apoptosis
7. Autophagy Mediated by HIV-1 Env
8. Targeting HIV gp41 Mediated Fusion Can Alter Bystander Apoptosis Inducing Function of Env
9. Immune Activation in HIV Disease Progression
10. Bystander Apoptosis in HIV Infected Individuals
11. Bystander Apoptosis in Animal Models of HIV Infection
12. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Hurtrel, B.; Petit, F.; Arnoult, D.; Muller-Trutwin, M.; Silvestri, G.; Estaquier, J. Apoptosis in SIV infection. Cell Death Differ. 2005, 12 Suppl. 1, 979–990. [Google Scholar] [CrossRef]
- Silvestri, G. AIDS pathogenesis: A tale of two monkeys. J. Med. Primatol. 2008, 37 Suppl. 2, 6–12. [Google Scholar] [CrossRef]
- Gougeon, M.; Montagnier, L. Apoptosis in AIDS. Science 1993, 260, 1269–1270. [Google Scholar]
- Laurent-Crawford, A.; Krust, B.; Rivière, Y.; Desgranges, C.; Muller, S.; Kieny, M.; Dauguet, C.; Hovanessian, A. Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res. Hum. Retroviruses 1993, 9, 761–773. [Google Scholar] [CrossRef]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barre-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; et al. Programmed cell death and AIDS: Significance of T-cell apoptosis in pathogenic and nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 1994, 91, 9431–9435. [Google Scholar]
- Finkel, T.; Tudor-Williams, G.; Banda, N.; Cotton, M.; Curiel, T.; Monks, C.; Baba, T.; Ruprecht, R.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Richard, V.; Roland, J.; Hipskind, R.; Devaux, C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120). Virology 2000, 268, 329–344. [Google Scholar] [CrossRef]
- Ferri, K.F.; Jacotot, E.; Geuskens, M.; Kroemer, G. Apoptosis and karyogamy in syncytia induced by the HIV-1-envelope glycoprotein complex. Cell Death Differ. 2000, 7, 1137–1139. [Google Scholar]
- Ahr, B.; Robert-Hebmann, V.; Devaux, C.; Biard-Piechaczyk, M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 2004, 1. [Google Scholar]
- Gougeon, M. To kill or be killed: How HIV exhausts the immune system. Cell Death Differ. 2005, 12 Suppl. 1, 845–854. [Google Scholar] [CrossRef]
- Douek, D.C.; Roederer, M.; Koup, R.A. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 2009, 60, 471–484. [Google Scholar]
- Finkel, T.; Banda, N. Indirect mechanisms of HIV pathogenesis: how does HIV kill T cells? Curr. Opin. Immunol. 1994, 6, 605–615. [Google Scholar] [CrossRef]
- Gougeon, M.; Colizzi, V.; Dalgleish, A.; Montagnier, L. New concepts in AIDS pathogenesis. AIDS Res. Hum. Retroviruses 1993, 9, 287–289. [Google Scholar] [CrossRef]
- Perfettini, J.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12 Suppl. 1, 916–923. [Google Scholar] [CrossRef]
- Garg, H.; Blumenthal, R. Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell. Mol. Life Sci. 2008, 65, 3134–3144. [Google Scholar] [CrossRef]
- Wyatt, R.; Sodroski, J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef]
- Gallo, S.; Finnegan, C.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 2003, 1614, 36–50. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar]
- Wild, C.; Dubay, J.; Greenwell, T.; Baird, T.J.; Oas, T.; McDanal, C.; Hunter, E.; Matthews, T. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12676–12680. [Google Scholar]
- Sourisseau, M.; Sol-Foulon, N.; Porrot, F.; Blanchet, F.; Schwartz, O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 2007, 81, 1000–1012. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar]
- Jolly, C.; Mitar, I.; Sattentau, Q. Adhesion molecule interactions facilitate human immunodeficiencyvirus type 1-induced virological synapse formation between T cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 2007, 81, 7873–7884. [Google Scholar] [CrossRef]
- Jolly, C.; Mitar, I.; Sattentau, Q. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 2007, 81, 5547–5560. [Google Scholar]
- Andreau, K.; Perfettini, J.; Castedo, M.; Métivier, D.; Scott, V.; Pierron, G.; Kroemer, G. Contagious apoptosis facilitated by the HIV-1 envelope: Fusion-induced cell-to-cell transmission of a lethal signal. J. Cell Sci. 2004, 117, 5643–5653. [Google Scholar] [CrossRef]
- Blanco, J.; Barretina, J.; Henson, G.; Bridger, G.; De Clercq, E.; Clotet, B.; Esté, J. The CXCR4 antagonist AMD3100 efficiently inhibits cell-surface-expressed human immunodeficiency virus type 1 envelope-induced apoptosis. Antimicrob. Agents Chemother. 2000, 44, 51–56. [Google Scholar] [CrossRef]
- Blanco, J.; Barretina, J.; Ferri, K.; Jacotot, E.; Gutiérrez, A.; Armand-Ugón, M.; Cabrera, C.; Kroemer, G.; Clotet, B.; Esté, J. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology 2003, 305, 318–329. [Google Scholar]
- Garg, H.; Joshi, A.; Freed, E.; Blumenthal, R. Site-specific mutations in HIV-1 gp41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J. Biol. Chem. 2007, 282, 16899–16906. [Google Scholar] [CrossRef]
- Meissner, E.; Zhang, L.; Jiang, S.; Su, L. Fusion-induced apoptosis contributes to thymocyte depletion by a pathogenic human immunodeficiency virus type 1 envelope in the human thymus. J. Virol. 2006, 80, 11019–11030. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Perfettini, J.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. NF-kappaB and p53 are the dominant apoptosis-inducing transcription factors elicited by the HIV-1 envelope. J. Exp. Med. 2004, 199, 629–640. [Google Scholar] [CrossRef]
- Spijkerman, I.; de Wolf, F.; Langendam, M.; Schuitemaker, H.; Coutinho, R. Emergence of syncytium-inducing human immunodeficiency virus type 1 variants coincides with a transient increase in viral RNA level and is an independent predictor for progression to AIDS. J. Infect. Dis. 1998, 178, 397–403. [Google Scholar] [CrossRef]
- Castedo, M.; Kroemer, G. Mitotic catastrophe: A special case of apoptosis. J. Soc. Biol. 2004, 198, 97–103. [Google Scholar]
- Roumier, T.; Castedo, M.; Perfettini, J.L.; Andreau, K.; Metivier, D.; Zamzami, N.; Kroemer, G. Mitochondrion-dependent caspase activation by the HIV-1 envelope. Biochem. Pharmacol. 2003, 66, 1321–1329. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Yakushijin, K.; Horne, D.; Medema, R.; Kroemer, G. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 2004, 23, 4353–4361. [Google Scholar] [CrossRef]
- Castedo, M.; Ferri, K.F.; Blanco, J.; Roumier, T.; Larochette, N.; Barretina, J.; Amendola, A.; Nardacci, R.; Metivier, D.; Este, J.A.; et al. Human immunodeficiency virus 1 envelope glycoprotein complex-induced apoptosis involves mammalian target of rapamycin/FKBP12-rapamycin-associated protein-mediated p53 phosphorylation. J. Exp. Med. 2001, 194, 1097–1110. [Google Scholar] [CrossRef]
- Ferri, K.F.; Kroemer, G. Control of apoptotic DNA degradation. Nat. Cell Biol. 2000, 2, E63–E64. [Google Scholar]
- Ferri, K.F.; Jacotot, E.; Blanco, J.; Este, J.A.; Zamzami, N.; Susin, S.A.; Xie, Z.; Brothers, G.; Reed, J.C.; Penninger, J.M.; et al. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: Role of mitochondria and caspases. J. Exp. Med. 2000, 192, 1081–1092. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Castedo, M.; Roumier, T.; Blanco, J.; Ferri, K.F.; Barretina, J.; Tintignac, L.A.; Andreau, K.; Perfettini, J.L.; Amendola, A.; Nardacci, R.; et al. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the HIV-1 envelope. EMBO J. 2002, 21, 4070–4080. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Castedo, M.; Nardacci, R.; Ciccosanti, F.; Boya, P.; Roumier, T.; Larochette, N.; Piacentini, M.; Kroemer, G. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J. Exp. Med. 2005, 201, 279–289. [Google Scholar] [CrossRef]
- Cannavo, G.; Paiardini, M.; Galati, D.; Cervasi, B.; Montroni, M.; De Vico, G.; Guetard, D.; Bocchino, M.L.; Picerno, I.; Magnani, M.; et al. Abnormal intracellular kinetics of cell-cycle-dependent proteins in lymphocytes from patients infected with human immunodeficiency virus: A novel biologic link between immune activation, accelerated T-cell turnover, and high levels of apoptosis. Blood 2001, 97, 1756–1764. [Google Scholar] [CrossRef]
- Piedimonte, G.; Corsi, D.; Paiardini, M.; Cannavo, G.; Ientile, R.; Picerno, I.; Montroni, M.; Silvestri, G.; Magnani, M. Unscheduled cyclin B expression and p34 cdc2 activation in T lymphocytes from HIV-infected patients. AIDS 1999, 13, 1159–1164. [Google Scholar] [CrossRef]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–287. [Google Scholar]
- Chernomordik, L.; Kozlov, M. Membrane hemifusion: Crossing a chasm in two leaps. Cell 2005, 123, 375–382. [Google Scholar]
- Bar, S.; Alizon, M. Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J. Virol. 2004, 78, 811–820. [Google Scholar] [CrossRef]
- Wang, X.M.; Nadeau, P.E.; Lo, Y.T.; Mergia, A. Caveolin-1 modulates HIV-1 envelope-induced bystander apoptosis through gp41. J. Virol. 2010, 84, 6515–6526. [Google Scholar] [CrossRef]
- Garg, H.; Blumenthal, R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J. Leukoc. Biol. 2006, 79, 351–362. [Google Scholar]
- Ashkenazi, A.; Viard, M.; Wexler-Cohen, Y.; Blumenthal, R.; Shai, Y. Viral envelope protein folding and membrane hemifusion are enhanced by the conserved loop region of HIV-1 gp41. FASEB J. 2011, 25, 2156–2166. [Google Scholar] [CrossRef]
- Barretina, J.; Blanco, J.; Armand-Ugon, M.; Gutierrez, A.; Clotet, B.; Este, J.A. Anti-HIV-1 activity of enfuvirtide (T-20) by inhibition of bystander cell death. Antivir. Ther. 2003, 8, 155–161. [Google Scholar]
- Cunyat, F.; Curriu, M.; Marfil, S.; Garcia, E.; Clotet, B.; Blanco, J.; Cabrera, C. Evaluation of the Cytopathicity (Fusion/Hemifusion) of Patient-Derived HIV-1 Envelope Glycoproteins Comparing Two Effector Cell Lines. J. Biomol. Screen. 2012, 17, 727–737. [Google Scholar] [CrossRef]
- Etemad-Moghadam, B.; Rhone, D.; Steenbeke, T.; Sun, Y.; Manola, J.; Gelman, R.; Fanton, J.; Racz, P.; Tenner-Racz, K.; Axthelm, M.; et al. Membrane-fusing capacity of the human immunodeficiency virus envelope proteins determines the efficiency of CD+ T-cell depletion in macaques infected by a simian-human immunodeficiency virus. J. Virol. 2001, 75, 5646–5655. [Google Scholar]
- Etemad-Moghadam, B.; Sun, Y.; Nicholson, E.; Fernandes, M.; Liou, K.; Gomila, R.; Lee, J.; Sodroski, J. Envelope glycoprotein determinants of increased fusogenicity in a pathogenic simian-human immunodeficiency virus (SHIV-KB9) passaged in vivo. J. Virol. 2000, 74, 4433–4440. [Google Scholar] [CrossRef]
- Koot, M.; van 't Wout, A.; Kootstra, N.; de Goede, R.; Tersmette, M.; Schuitemaker, H. Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection. J. Infect. Dis. 1996, 173, 349–354. [Google Scholar] [CrossRef]
- Schuitemaker, H.; Koot, M.; Kootstra, N.; Dercksen, M.; de Goede, R.; van Steenwijk, R.; Lange, J.; Schattenkerk, J.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: Progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar]
- van Rij, R.P.; Blaak, H.; Visser, J.; Brouwer, M.; Rientsma, R.; Broersen, S.; de Roda Husman, A.M.; Schuitemaker, H. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J. Clin. Invest. 2000, 106, 1569. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A.; Ye, C.; Shankar, P.; Manjunath, N. Single amino acid change in gp41 region of HIV-1 alters bystander apoptosis and CD4 decline in humanized mice. Virol. J. 2011, 8. [Google Scholar]
- Jekle, A.; Keppler, O.; De Clercq, E.; Schols, D.; Weinstein, M.; Goldsmith, M. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J. Virol. 2003, 77, 5846–5854. [Google Scholar] [CrossRef]
- Anton, P.A.; Elliott, J.; Poles, M.A.; McGowan, I.M.; Matud, J.; Hultin, L.E.; Grovit-Ferbas, K.; Mackay, C.R.; Chen, I.S.Y.; Giorgi, J.V. Enhanced levels of functional HIV-1 co-receptors on human mucosal T cells demonstrated using intestinal biopsy tissue. AIDS 2000, 14, 1761–1765. [Google Scholar]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef]
- Sterjovski, J.; Churchill, M.J.; Ellett, A.; Gray, L.R.; Roche, M.J.; Dunfee, R.L.; Purcell, D.F.; Saksena, N.; Wang, B.; Sonza, S.; et al. Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology 2007, 4. [Google Scholar]
- Holm, G.; Zhang, C.; Gorry, P.; Peden, K.; Schols, D.; De Clercq, E.; Gabuzda, D. Apoptosis of bystander T cells induced by human immunodeficiency virus type 1 with increased envelope/ receptor affinity and coreceptor binding site exposure. J. Virol. 2004, 78, 4541–4551. [Google Scholar] [CrossRef]
- Wade, J.; Sterjovski, J.; Gray, L.; Roche, M.; Chiavaroli, L.; Ellett, A.; Jakobsen, M.R.; Cowley, D.; Pereira Cda, F.; Saksena, N.; et al. Enhanced CD4+ cellular apoptosis by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with progressive HIV-1 infection. Virology 396, 246–255.
- Olivieri, K.; Scoggins, R.; Bor, Y.; Matthews, A.; Mark, D.; Taylor, J.J.; Chernauskas, D.; Hammarskjöld, M.; Rekosh, D.; Camerini, D. The envelope gene is a cytopathic determinant of CCR5 tropic HIV-1. Virology 2007, 358, 23–38. [Google Scholar]
- Bjorndal, A.; Sonnerborg, A.; Tscherning, C.; Albert, J.; Fenyo, E.M. Phenotypic characteristics of human immunodeficiency virus type 1 subtype C isolates of Ethiopian AIDS patients. AIDS Res. Hum. Retroviruses 1999, 15, 647–653. [Google Scholar] [CrossRef]
- Edo-Matas, D.; van Dort, K.A.; Setiawan, L.C.; Schuitemaker, H.; Kootstra, N.A. Comparison of in vivo and in vitro evolution of CCR5 to CXCR4 coreceptor use of primary human immunodeficiency virus type 1 variants. Virology 2011, 412, 269–277. [Google Scholar] [CrossRef]
- Huang, W.; Eshleman, S.H.; Toma, J.; Fransen, S.; Stawiski, E.; Paxinos, E.E.; Whitcomb, J.M.; Young, A.M.; Donnell, D.; Mmiro, F.; et al. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: High prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J. Virol. 2007, 81, 7885–7893. [Google Scholar]
- Coetzer, M.; Nedellec, R.; Cilliers, T.; Meyers, T.; Morris, L.; Mosier, D.E. Extreme genetic divergence is required for coreceptor switching in HIV-1 subtype C. J. Acquir. Immune Defic. Syndr. 2011, 56, 9–15. [Google Scholar] [CrossRef]
- Ng, O.T.; Lin, L.; Laeyendecker, O.; Quinn, T.C.; Sun, Y.J.; Lee, C.C.; Leo, Y.S. Increased rate of CD4+ T-cell decline and faster time to antiretroviral therapy in HIV-1 subtype CRF01_AE infected seroconverters in Singapore. PLoS One 2011, 6, e15738. [Google Scholar]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.; Choe, S.; Ceradini, D.; Martin, S.; Horuk, R.; MacDonald, M.; Stuhlmann, H.; Koup, R.; Landau, N. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Marmor, M.; Sheppard, H.; Donnell, D.; Bozeman, S.; Celum, C.; Buchbinder, S.; Koblin, B.; Seage, G.R. Homozygous and heterozygous CCR5-Delta32 genotypes are associated with resistance to HIV infection. J. Acquir. Immune Defic. Syndr. 2001, 27, 472–481. [Google Scholar]
- de Roda Husman, A.M.; Koot, M.; Cornelissen, M.; Keet, I.P.; Brouwer, M.; Broersen, S.M.; Bakker, M.; Roos, M.T.; Prins, M.; de Wolf, F.; et al. Association between CCR5 genotype and the clinical course of HIV-1 infection. Ann. Intern. Med. 1997, 127, 882–890. [Google Scholar]
- Ometto, L.; Bertorelle, R.; Mainardi, M.; Zanchetta, M.; Tognazzo, S.; Rampon, O.; Ruga, E.; Chieco-Bianchi, L.; De Rossi, A. Polymorphisms in the CCR5 promoter region influence disease progression in perinatally human immunodeficiency virus type 1-infected children. J. Infect. Dis. 2001, 183, 814–818. [Google Scholar] [CrossRef]
- Shieh, B.; Liau, Y.; Hsieh, P.; Yan, Y.; Wang, S.; Li, C. Influence of nucleotide polymorphisms in the CCR2 gene and the CCR5 promoter on the expression of cell surface CCR5 and CXCR4. Int. Immunol. 2000, 12, 1311–1318. [Google Scholar]
- Reeves, J.; Gallo, S.; Ahmad, N.; Miamidian, J.; Harvey, P.; Sharron, M.; Pohlmann, S.; Sfakianos, J.; Derdeyn, C.; Blumenthal, R.; et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16249–16254. [Google Scholar]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997, 185, 1681–1691. [Google Scholar] [CrossRef]
- Scoggins, R.; Taylor, J.J.; Patrie, J.; van't Wout, A.; Schuitemaker, H.; Camerini, D. Pathogenesis of primary R5 human immunodeficiency virus type 1 clones in SCID-hu mice. J. Virol. 2000, 74, 3205–3216. [Google Scholar]
- Paiardini, M.; Cervasi, B.; Reyes-Aviles, E.; Micci, L.; Ortiz, A.M.; Chahroudi, A.; Vinton, C.; Gordon, S.N.; Bosinger, S.E.; Francella, N.; et al. Low levels of SIV infection in sooty mangabey central memory CD(4)(+) T cells are associated with limited CCR5 expression. Nat. Med. 2011, 17, 830–836. [Google Scholar]
- Platt, E.; Wehrly, K.; Kuhmann, S.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar]
- Platt, E.; Durnin, J.; Kabat, D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 2005, 79, 4347–4356. [Google Scholar] [CrossRef]
- Joshi, A.; Nyakeriga, A.M.; Ravi, R.; Garg, H. HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J. Biol. Chem. 2011, 286, 36404–36413. [Google Scholar]
- Schmid, D.; Munz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Espert, L.; Biard-Piechaczyk, M. Autophagy in HIV-induced T cell death. Curr. Top. Microbiol. Immunol. 2009, 335, 307–321. [Google Scholar]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One 2009, 4, e5787. [Google Scholar]
- Spector, S.A.; Zhou, D. Autophagy: An overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy 2008, 4, 704–706. [Google Scholar]
- Deretic, V. Autophagy as an immune defense mechanism. Curr. Opin. Immunol. 2006, 18, 375–382. [Google Scholar]
- Deretic, V. Autophagy of intracellular microbes and mitochondria: Two sides of the same coin? F1000 Biol. Rep. 2010, 2. [Google Scholar]
- Deretic, V. Autophagy in infection. Curr. Opin. Cell Biol. 2010, 22, 252–262. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Gougeon, M.L.; Piacentini, M. New insights on the role of apoptosis and autophagy in HIV pathogenesis. Apoptosis 2009, 14, 501–508. [Google Scholar] [CrossRef]
- Wild, C.; Shugars, D.; Greenwell, T.; McDanal, C.; Matthews, T. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef]
- Reeves, J.; Lee, F.; Miamidian, J.; Jabara, C.; Juntilla, M.; Doms, R. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 2005, 79, 4991–4999. [Google Scholar] [CrossRef]
- Barretina, J.; Blanco, J.; Bonjoch, A.; Llano, A.; Clotet, B.; Esté, J. Immunological and virological study of enfuvirtide-treated HIV-positive patients. AIDS 2004, 18, 1673–1682. [Google Scholar]
- Bonora, S.; Calcagno, A.; Cometto, C.; Fontana, S.; Aguilar, D.; D'Avolio, A.; Gonzalez de Requena, D.; Maiello, A.; Dal Conte, I.; Lucchini, A.; et al. Short-term additional enfuvirtide therapy is associated with a greater immunological recovery in HIV very late presenters: A controlled pilot study. Infection 2012, 40, 69–75. [Google Scholar] [CrossRef]
- Aquaro, S.; D'Arrigo, R.; Svicher, V.; Perri, G.; Caputo, S.; Visco-Comandini, U.; Santoro, M.; Bertoli, A.; Mazzotta, F.; Bonora, S.; et al. Specific mutations in HIV-1 gp41 are associated with immunological success in HIV-1-infected patients receiving enfuvirtide treatment. J. Antimicrob. Chemother. 2006, 58, 714–722. [Google Scholar] [CrossRef]
- Melby, T.; Despirito, M.; Demasi, R.; Heilek, G.; Thommes, J.; Greenberg, M.; Graham, N. Association between specific enfuvirtide resistance mutations and CD4 cell response during enfuvirtide-based therapy. AIDS 2007, 21, 2537–2539. [Google Scholar] [CrossRef]
- Svicher, V.; Aquaro, S.; D'Arrigo, R.; Artese, A.; Dimonte, S.; Alcaro, S.; Santoro, M.; Di Perri, G.; Caputo, S.; Bellagamba, R.; et al. Specific enfuvirtide-associated mutational pathways in HIV-1 Gp41 are significantly correlated with an increase in CD4(+) cell count, despite virological failure. J. Infect. Dis. 2008, 197, 1408–1418. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A.; Blumenthal, R. Altered bystander apoptosis induction and pathogenesis of enfuvirtide-resistant HIV type 1 Env mutants. AIDS Res. Hum. Retroviruses 2009, 25, 811–817. [Google Scholar]
- Cunyat, F.; Marfil, S.; Garcia, E.; Svicher, V.; Perez-Alvarez, N.; Curriu, M.; Perno, C.F.; Clotet, B.; Blanco, J.; Cabrera, C. The HR2 polymorphism N140I in the HIV-1 gp41 combined with the HR1 V38A mutation is associated with a less cytopathic phenotype. Retrovirology 2012, 9. [Google Scholar]
- Bentwich, Z.; Kalinkovich, A.; Weisman, Z.; Grossman, Z. Immune activation in the context of HIV infection. Clin. Exp. Immunol. 1998, 111, 1–2. [Google Scholar]
- Sodora, D.; Silvestri, G. Immune activation and AIDS pathogenesis. AIDS 2008, 22, 439–446. [Google Scholar] [CrossRef]
- Estes, J.; Gordon, S.; Zeng, M.; Chahroudi, A.; Dunham, R.; Staprans, S.; Reilly, C.; Silvestri, G.; Haase, A. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J. Immunol. 2008, 180, 6798–6807. [Google Scholar]
- Silvestri, G.; Sodora, D.; Koup, R.; Paiardini, M.; O'Neil, S.; McClure, H.; Staprans, S.; Feinberg, M. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003, 18, 441–452. [Google Scholar]
- Grossman, Z.; Meier-Schellersheim, M.; Paul, W.; Picker, L. Pathogenesis of HIV infection: What the virus spares is as important as what it destroys. Nat. Med. 2006, 12, 289–295. [Google Scholar] [CrossRef]
- Grossman, Z.; Meier-Schellersheim, M.; Sousa, A.; Victorino, R.; Paul, W. CD4+ T-cell depletion in HIV infection: Are we closer to understanding the cause? Nat. Med. 2002, 8, 319–323. [Google Scholar] [CrossRef]
- Al-Harthi, L.; MaWhinney, S.; Connick, E.; Schooley, R.; Forster, J.; Benson, C.; Thompson, M.; Judson, F.; Palella, F.; Landay, A. Immunophenotypic alterations in acute and early HIV infection. Clin. Immunol. 2007, 125, 299–308. [Google Scholar] [CrossRef]
- Biancotto, A.; Grivel, J.; Iglehart, S.; Vanpouille, C.; Lisco, A.; Sieg, S.; Debernardo, R.; Garate, K.; Rodriguez, B.; Margolis, L.; et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood 2007, 109, 4272–4279. [Google Scholar] [CrossRef]
- Gougeon, M. T cell apoptosis as a consequence of chronic activation of the immune system in HIV infection. Adv. Exp. Med. Biol. 1995, 374, 121–127. [Google Scholar]
- Leng, Q.; Borkow, G.; Weisman, Z.; Stein, M.; Kalinkovich, A.; Bentwich, Z. Immune activation correlates better than HIV plasma viral load with CD4 T-cell decline during HIV infection. J. Acquir. Immune Defic. Syndr. 2001, 27, 389–397. [Google Scholar]
- Sousa, A.; Carneiro, J.; Meier-Schellersheim, M.; Grossman, Z.; Victorino, R. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 2002, 169, 3400–3406. [Google Scholar]
- Resino, S.; Seoane, E.; Gutiérrez, M.; León, J.; Muñoz-Fernández, M. CD4(+) T-cell immunodeficiency is more dependent on immune activation than viral load in HIV-infected children on highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2006, 42, 269–276. [Google Scholar] [CrossRef]
- Biancotto, A.; Iglehart, S.; Vanpouille, C.; Condack, C.; Lisco, A.; Ruecker, E.; Hirsch, I.; Margolis, L.; Grivel, J. HIV-1 induced activation of CD4+ T cells creates new targets for HIV-1 infection in human lymphoid tissue ex vivo. Blood 2008, 111, 699–704. [Google Scholar] [CrossRef]
- Koning, F.; Otto, S.; Hazenberg, M.; Dekker, L.; Prins, M.; Miedema, F.; Schuitemaker, H. Low-level CD4+ T cell activation is associated with low susceptibility to HIV-1 infection. J. Immunol. 2005, 175, 6117–6122. [Google Scholar]
- Kaufmann, G.; Zaunders, J.; Cooper, D. Immune reconstitution in HIV-1 infected subjects treated with potent antiretroviral therapy. Sex. Transm. Infect. 1999, 75, 218–224. [Google Scholar] [CrossRef]
- Brenchley, J.; Price, D.; Schacker, T.; Asher, T.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar]
- Redd, A.D.; Gray, R.H.; Quinn, T.C. Is microbial translocation a cause or consequence of HIV disease progression? J. Infect. Dis. 2011, 203, 744–745, author reply 746.. [Google Scholar] [CrossRef]
- Brainard, D.M.; Seung, E.; Frahm, N.; Cariappa, A.; Bailey, C.C.; Hart, W.K.; Shin, H.S.; Brooks, S.F.; Knight, H.L.; Eichbaum, Q.; et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol. 2009, 83, 7305–7321. [Google Scholar]
- Ameisen, J.C.; Capron, A. Cell dysfunction and depletion in AIDS: The programmed cell death hypothesis. Immunol. Today 1991, 12, 102–105. [Google Scholar] [CrossRef]
- Lecoeur, H.; Gougeon, M.L. Comparative analysis of flow cytometric methods for apoptosis quantitation in murine thymocytes and human peripheral lymphocytes from controls and HIV-infected persons. Evidence for interference by granulocytes and erythrocytes. J. Immunol. Methods 1996, 198, 87–99. [Google Scholar] [CrossRef]
- Peraire, J.; Miro, O.; Saumoy, M.; Domingo, P.; Pedrol, E.; Villarroya, F.; Martinez, E.; Lopez-Dupla, M.; Garrabou, G.; Sambeat, M.A.; et al. HIV-1-infected long-term non-progressors have milder mitochondrial impairment and lower mitochondrially-driven apoptosis in peripheral blood mononuclear cells than typical progressors. Curr. HIV Res. 2007, 5, 467–473. [Google Scholar] [CrossRef]
- Sternfeld, T.; Tischleder, A.; Schuster, M.; Bogner, J.R. Mitochondrial membrane potential and apoptosis of blood mononuclear cells in untreated HIV-1 infected patients. HIV Med. 2009, 10, 512–519. [Google Scholar]
- Badley, A.; Roumier, T.; Lum, J.; Kroemer, G. Mitochondrion-mediated apoptosis in HIV-1 infection. Trends Pharmacol. Sci. 2003, 24, 298–305. [Google Scholar]
- de Oliveira Pinto, L.M.; Garcia, S.; Lecoeur, H.; Rapp, C.; Gougeon, M.L. Increased sensitivity of T lymphocytes to tumor necrosis factor receptor 1 (TNFR1)- and TNFR2-mediated apoptosis in HIV infection: Relation to expression of Bcl-2 and active caspase-8 and caspase-3. Blood 2002, 99, 1666–1675. [Google Scholar] [CrossRef]
- Rey-Cuille, M.A.; Berthier, J.L.; Bomsel-Demontoy, M.C.; Chaduc, Y.; Montagnier, L.; Hovanessian, A.G.; Chakrabarti, L.A. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J. Virol. 1998, 72, 3872–3886. [Google Scholar]
- Chakrabarti, L.A.; Lewin, S.R.; Zhang, L.; Gettie, A.; Luckay, A.; Martin, L.N.; Skulsky, E.; Ho, D.D.; Cheng-Mayer, C.; Marx, P.A. Normal T-cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J. Virol. 2000, 74, 1209–1223. [Google Scholar]
- Cumont, M.C.; Diop, O.; Vaslin, B.; Elbim, C.; Viollet, L.; Monceaux, V.; Lay, S.; Silvestri, G.; Le Grand, R.; Muller-Trutwin, M.; et al. Early divergence in lymphoid tissue apoptosis between pathogenic and nonpathogenic simian immunodeficiency virus infections of nonhuman primates. J. Virol. 2008, 82, 1175–1184. [Google Scholar]
- Meythaler, M.; Martinot, A.; Wang, Z.; Pryputniewicz, S.; Kasheta, M.; Ling, B.; Marx, P.A.; O'Neil, S.; Kaur, A. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J. Virol. 2009, 83, 572–583. [Google Scholar]
- Meythaler, M.; Pryputniewicz, S.; Kaur, A. Kinetics of T lymphocyte apoptosis and the cellular immune response in SIVmac239-infected rhesus macaques. J. Med. Primatol. 2008, 37 Suppl. 2, 33–45. [Google Scholar] [CrossRef]
- Reimann, K.A.; Li, J.T.; Veazey, R.; Halloran, M.; Park, I.W.; Karlsson, G.B.; Sodroski, J.; Letvin, N.L. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J. Virol. 1996, 70, 6922–6928. [Google Scholar]
- Karlsson, G.B.; Halloran, M.; Li, J.; Park, I.W.; Gomila, R.; Reimann, K.A.; Axthelm, M.K.; Iliff, S.A.; Letvin, N.L.; Sodroski, J. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J. Virol. 1997, 71, 4218–4225. [Google Scholar]
- Karlsson, G.; Halloran, M.; Schenten, D.; Lee, J.; Racz, P.; Tenner-Racz, K.; Manola, J.; Gelman, R.; Etemad-Moghadam, B.; Desjardins, E.; et al. The envelope glycoprotein ectodomains determine the efficiency of CD4+ T lymphocyte depletion in simian-human immunodeficiency virus-infected macaques. J. Exp. Med. 1998, 188, 1159–1171. [Google Scholar] [CrossRef]
- LaBonte, J.; Patel, T.; Hofmann, W.; Sodroski, J. Importance of membrane fusion mediated by human immunodeficiency virus envelope glycoproteins for lysis of primary CD4-positive T cells. J. Virol. 2000, 74, 10690–10698. [Google Scholar] [CrossRef]
- Berges, B.K.; Rowan, M.R. The utility of the new generation of humanized mice to study HIV-1 infection: Transmission, prevention, pathogenesis, and treatment. Retrovirology 2011, 8. [Google Scholar]
- Berges, B.; Wheat, W.; Palmer, B.; Connick, E.; Akkina, R. HIV-1 infection and CD4 T cell depletion in the humanized Rag2−/− gamma c−/− (RAG-hu) mouse model. Retrovirology 2006, 3. [Google Scholar]
- Shultz, L.; Lang, P.; Christianson, S.; Gott, B.; Lyons, B.; Umeda, S.; Leiter, E.; Hesselton, R.; Wagar, E.; Leif, J.; et al. NOD/LtSz-Rag1null mice: An immunodeficient and radioresistant model for engraftment of human hematolymphoid cells, HIV infection, and adoptive transfer of NOD mouse diabetogenic T cells. J. Immunol. 2000, 164, 2496–2507. [Google Scholar]
- Zhang, L.; Kovalev, G.; Su, L. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood 2007, 109, 2978–2981. [Google Scholar]
- Baenziger, S.; Tussiwand, R.; Schlaepfer, E.; Mazzucchelli, L.; Heikenwalder, M.; Kurrer, M.; Behnke, S.; Frey, J.; Oxenius, A.; Joller, H.; et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2−/− gamma c−/− mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15951–15956. [Google Scholar]
- Camerini, D.; Su, H.; Gamez-Torre, G.; Johnson, M.; Zack, J.; Chen, I. Human immunodeficiency virus type 1 pathogenesis in SCID-hu mice correlates with syncytium-inducing phenotype and viral replication. J. Virol. 2000, 74, 3196–3204. [Google Scholar]
- Joshi, A.; Garg, H.; Ablan, S.; Freed, E.O.; Nagashima, K.; Manjunath, N.; Shankar, P. Targeting the HIV entry, assembly and release pathways for anti-HIV gene therapy. Virology 2011, 415, 95–106. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Garg, H.; Mohl, J.; Joshi, A. HIV-1 Induced Bystander Apoptosis. Viruses 2012, 4, 3020-3043. https://doi.org/10.3390/v4113020
Garg H, Mohl J, Joshi A. HIV-1 Induced Bystander Apoptosis. Viruses. 2012; 4(11):3020-3043. https://doi.org/10.3390/v4113020
Chicago/Turabian StyleGarg, Himanshu, Jonathon Mohl, and Anjali Joshi. 2012. "HIV-1 Induced Bystander Apoptosis" Viruses 4, no. 11: 3020-3043. https://doi.org/10.3390/v4113020
APA StyleGarg, H., Mohl, J., & Joshi, A. (2012). HIV-1 Induced Bystander Apoptosis. Viruses, 4(11), 3020-3043. https://doi.org/10.3390/v4113020