Involvement of Autophagy in Coronavirus Replication

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Coronavirus Genome Transcription and Replication

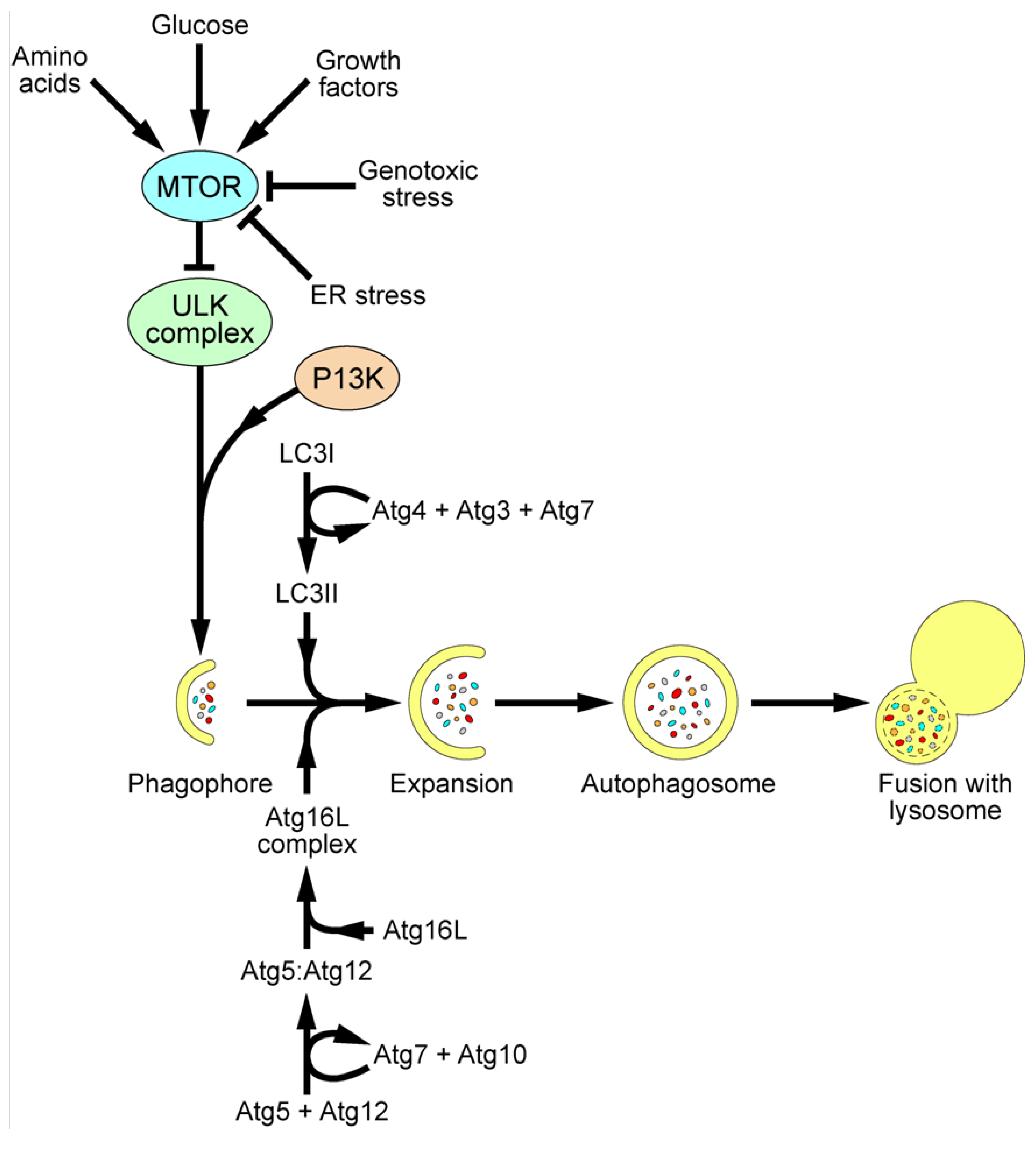
3. Autophagy

5. Future Questions and Perspectives
Acknowledgements
Conflict of Interest
References and Notes
- Lai, M.M.C.; Perlman, S.; Anderson, L.J. Coronaviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philidelphia, PA, USA, 2007; pp. 1305–1327. [Google Scholar]
- Posthuma, C.C.; Pedersen, K.W.; Lu, Z.; Joosten, R.G.; Roos, N.; Zevenhoven-Dobbe, J.C.; Snijder, E.J. Formation of the arterivirus replication/transcription complex: A key role for nonstructural protein 3 in the remodeling of intracellular membranes. J. Virol. 2008, 82, 4480–4491. [Google Scholar]
- Snijder, E.J.; van Tol, H.; Roos, N.; Pedersen, K.W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 2001, 82, 985–994. [Google Scholar]
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef]
- Clementz, M.A.; Kanjanahaluethai, A.; O'Brien, T.E.; Baker, S.C. Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology 2008, 375, 118–129. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Ulasli, M.; Vonk, A.M.; Reggiori, F.; Rottier, P.J.M.; de Haan, C.A.M. Mobility and interactions of coronavirus nonstructural protein 4. J. Virol. 2011, 85, 4572–4577. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.M.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.M.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Vonk, A.M.; Monastyrska, I.; Rottier, P.J.M.; de Haan, C.A.M. Visualizing coronavirus rna synthesis in time by using click chemistry. J. Virol. 2012, 86, 5808–5816. [Google Scholar]
- Ding, W.X.; Yin, X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2007, 4, 141–150. [Google Scholar]
- Kapahi, P.; Chen, D.; Rogers, A.N.; Katewa, S.D.; Li, P.W.-L.; Thomas, E.L.; Kockel, L. With tor, less is more: A key role for the conserved nutrient-sensing tor pathway in aging. Cell Metab. 2010, 11, 453–465. [Google Scholar] [CrossRef]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Yoshimori, T. Autophagy: A regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004, 313, 453–458. [Google Scholar] [CrossRef]
- Zhou, S.; Zhao, L.; Kuang, M.; Zhang, B.; Liang, Z.; Yi, T.; Wei, Y.; Zhao, X. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012, 323, 115–127. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Ann. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5757. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in mhc-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef]
- Gannage, M.; Münz, C. Mhc presentation via autophagy and how viruses escape from it. Semin. Immunopathol. 2010, 32, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Tey, S.-K.; Khanna, R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 2012, 120, 994–1004. [Google Scholar] [CrossRef]
- Into, T.; Inomata, M.; Takayama, E.; Takigawa, T. Autophagy in regulation of toll-like receptor signaling. Cell. Signal. 2012, 24, 1150–1162. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy protects against sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Cherry, S. VSV infection is sensed by Drosophila, attenuates nutrient signaling, and thereby activates antiviral autophagy. Autophagy 2009, 5, 1062–1063. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Hong, H.S.; E, X.; Kim, K.S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral Bcl-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef]
- Lee, J.-S.; Li, Q.; Lee, J.-Y.; Lee, S.-H.; Jeong, J.H.; Lee, H.-R.; Chang, H.; Zhou, F.-C.; Gao, S.-J.; Liang, C.; et al. Flip-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with Hiv-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011, 85, 6319–6333. [Google Scholar] [CrossRef]
- Taylor, M.; Kirkegaard, K. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 2008, 4, 286–289. [Google Scholar]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Li, J.-K.; Liang, J.-J.; Liao, C.-L.; Lin, Y.-L. Autophagy is involved in the early step of japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Cottam, E.; Pierini, R.; Roberts, R.; Wileman, T. Origins of membrane vesicles generated during replication of positive-strand RNA viruses. Future Virol. 2009, 4, 473–485. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Levine, B. Selective autophagy and viruses. Autophagy 2011, 7, 260–265. [Google Scholar] [CrossRef]
- Dreux, M.; Chisari, F.V. Impact of the autophagy machinery on hepatitis C virus infection. Viruses 2011, 3, 1342–1357. [Google Scholar] [CrossRef]
- Tang, S.W.; Ducroux, A.; Jeang, K.T.; Neuveut, C. Impact of cellular autophagy on viruses: Insights from hepatitis B virus and human retroviruses. J. Biomed. Sci. 2012, 19. [Google Scholar] [CrossRef]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef]
- Orvedahl, A.; Levine, B. Viral evasion of autophagy. Autophagy 2007, 4, 280–285. [Google Scholar]
- Taylor, M.P.; Jackson, W.T. Viruses and arrested autophagosome development. Autophagy 2009, 5, 870–871. [Google Scholar]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W.T. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef]
- de Haan, C.A.; Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2007, 4, 276–279. [Google Scholar]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.M.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef]
- Bernasconi, R.; Galli, C.; Noack, J.; Bianchi, S.; de Haan, Cornelis A.M.; Reggiori, F.; Molinari, M. Role of the SEL1L:LC3-I complex as an ERAD tuning receptor in the mammalian ER. Mol. Cell 2012, 46, 809–819. [Google Scholar] [CrossRef]
- Bernasconi, R.; Noack, J.; Molinari, M. Unconventional roles of nonlipidated LC3 in ERAD tuning and coronavirus infection. Autophagy 2012, 8, 1534–1536. [Google Scholar]
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef]
- Calì, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maier, H.J.; Britton, P. Involvement of Autophagy in Coronavirus Replication. Viruses 2012, 4, 3440-3451. https://doi.org/10.3390/v4123440
Maier HJ, Britton P. Involvement of Autophagy in Coronavirus Replication. Viruses. 2012; 4(12):3440-3451. https://doi.org/10.3390/v4123440
Chicago/Turabian StyleMaier, Helena J., and Paul Britton. 2012. "Involvement of Autophagy in Coronavirus Replication" Viruses 4, no. 12: 3440-3451. https://doi.org/10.3390/v4123440
APA StyleMaier, H. J., & Britton, P. (2012). Involvement of Autophagy in Coronavirus Replication. Viruses, 4(12), 3440-3451. https://doi.org/10.3390/v4123440