Large Animal Models for Foamy Virus Vector Gene Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Advantages of Large Animal Models for HSC Gene Therapy
3. Canine Studies of FV HSC Gene Therapy
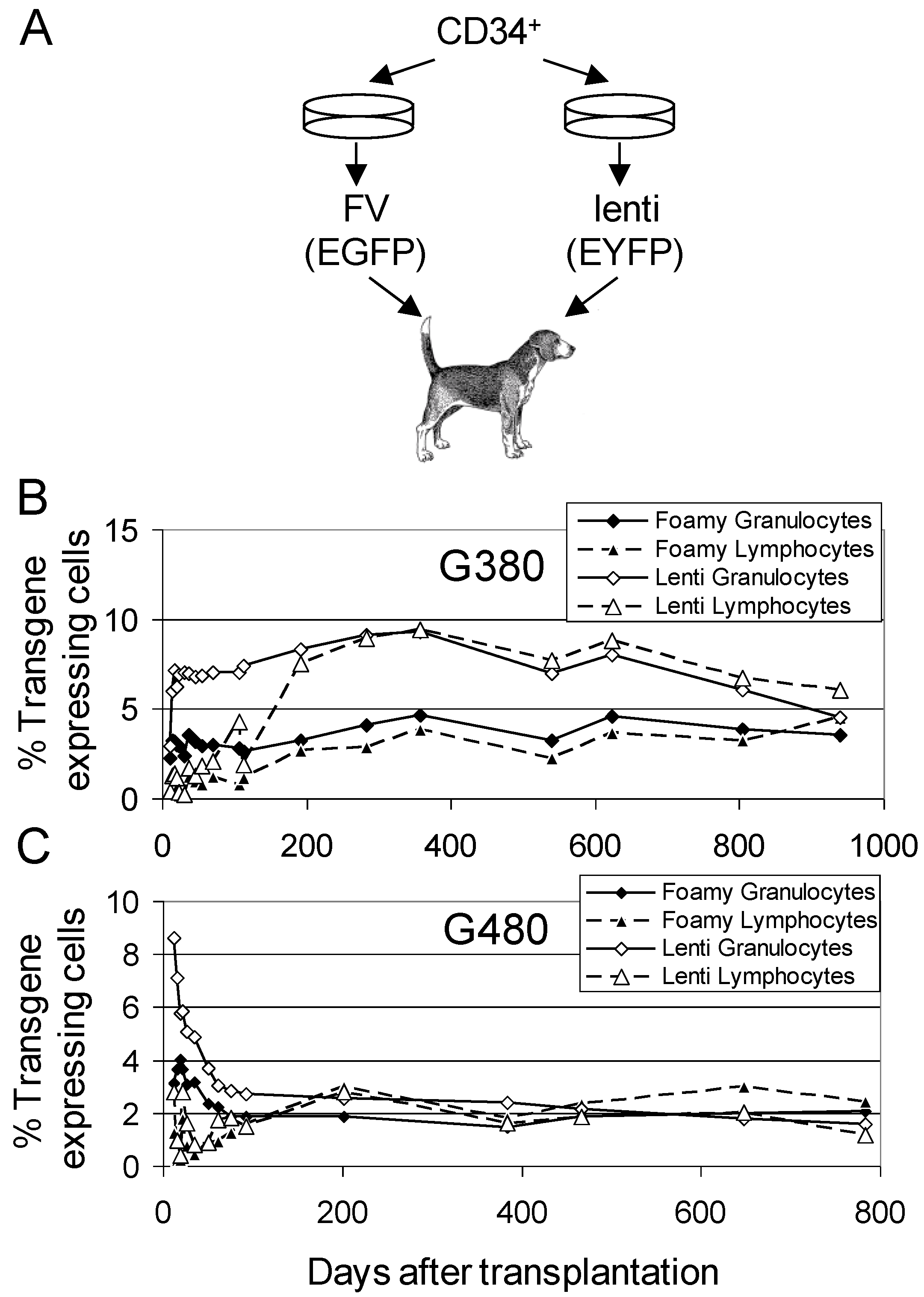
3.1. Efficient Multi-Lineage Gene Transfer Using FV Vectors
3.2. Comparison of FV Vectors to Lentiviral Vectors

3.3. FV Correction of Canine Leukocyte Adhesion Deficiency (CLAD)
3.4. Correction of Canine Pyruvate Kinase (PK) Deficiency
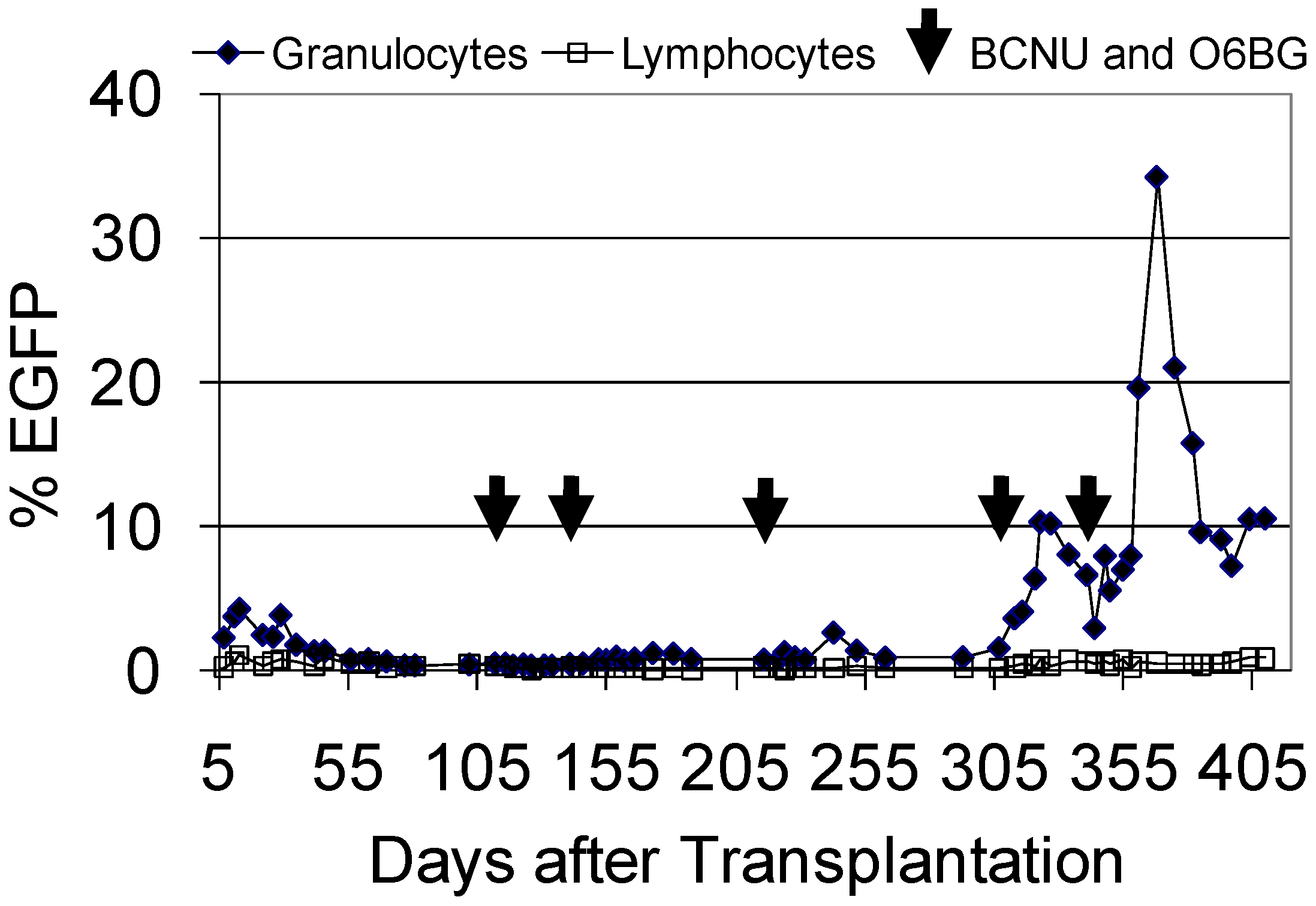
3.5. Evaluation of FV Genotoxicity in the Dog Large Animal Model
4. Nonhuman Primate Studies of FV Vector HSC Gene Therapy
4.1. Challenges Using Nonhuman Primate Models for FV HSC Gene Therapy

4.2. FV Gene Therapy in the Pigtailed Macaque

4.3. Progress in the Marmoset Model
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef]
- Linial, M. Why aren't foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Hill, C.L.; Bieniasz, P.D.; McClure, M.O. Properties of human foamy virus relevant to its development as a vector for gene therapy. J. Gen. Virol. 1999, 80, 2003–2009. [Google Scholar]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef]
- Josephson, N.C.; Trobridge, G.; Russell, D.W. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum. Gene Ther. 2004, 15, 87–92. [Google Scholar] [CrossRef]
- Josephson, N.C.; Vassilopoulos, G.; Trobridge, G.D.; Priestley, G.V.; Wood, B.L.; Papayannopoulou, T.; Russell, D.W. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc. Natl. Acad. Sci. USA 2002, 99, 8295–8300. [Google Scholar]
- Vassilopoulos, G.; Trobridge, G.; Josephson, N.C.; Russell, D.W. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 2001, 98, 604–609. [Google Scholar] [CrossRef]
- Leurs, C.; Jansen, M.; Pollok, K.E.; Heinkelein, M.; Schmidt, M.; Wissler, M.; Lindemann, D.; Von Kalle, C.; Rethwilm, A.; Williams, D.A.; Hanenberg, H. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34(+) cord blood cells. Hum. Gene Ther. 2003, 14, 509–519. [Google Scholar] [CrossRef]
- Si, Y.; Pulliam, A.C.; Linka, Y.; Ciccone, S.; Leurs, C.; Yuan, J.; Eckermann, O.; Fruehauf, S.; Mooney, S.; Hanenberg, H.; Clapp, D.W. Overnight transduction with foamyviral vectors restores the long-term repopulating activity of Fancc-/- stem cells. Blood 2008, 112, 4458–4465. [Google Scholar] [CrossRef]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar]
- Trobridge, G.D.; Kiem, H.P. Large animal models of hematopoietic stem cell gene therapy. Gene Ther. 2010, 17, 939–948. [Google Scholar] [CrossRef]
- Abkowitz, J.L.; Persik, M.T.; Shelton, G.H.; Ott, R.L.; Kiklevich, J.V.; Catlin, S.N.; Guttorp, P. Behavior of hematopoietic stem cells in a large animal. Proc. Natl. Acad. Sci. USA 1995, 92, 2031–2035. [Google Scholar]
- Horn, P.A.; Morris, J.C.; Neff, T.; Kiem, H.P. Stem cell gene transfer—Efficacy and safety in large animal studies. Mol Ther 2004, 10, 417–431. [Google Scholar] [CrossRef]
- Patton, G.S.; Erlwein, O.; McClure, M.O. Cell-cycle dependence of foamy virus vectors. J. Gen Virol. 2004, 85, 2925–2930. [Google Scholar] [CrossRef]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef]
- Abkowitz, J.L.; Golinelli, D.; Harrison, D.E.; Guttorp, P. In vivo kinetics of murine hemopoietic stem cells. Blood 2000, 96, 3399–3405. [Google Scholar]
- Shepherd, B.E.; Kiem, H.P.; Lansdorp, P.M.; Dunbar, C.E.; Aubert, G.; LaRochelle, A.; Seggewiss, R.; Guttorp, P.; Abkowitz, J.L. Hematopoietic stem-cell behavior in nonhuman primates. Blood 2007, 110, 1806–1813. [Google Scholar] [CrossRef]
- Shepherd, B.E.; Guttorp, P.; Lansdorp, P.M.; Abkowitz, J.L. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Exp. Hematol. 2004, 32, 1040–1050. [Google Scholar] [CrossRef]
- Gothot, A.; van der Loo, J.C.; Clapp, D.W.; Srour, E.F. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood 1998, 92, 2641–2649. [Google Scholar]
- Tisdale, J.F.; Hanazono, Y.; Sellers, S.E.; Agricola, B.A.; Metzger, M.E.; Donahue, R.E.; Dunbar, C.E. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood 1998, 92, 1131–1141. [Google Scholar]
- Young, J.C.; Lin, K.; Travis, M.; Hansteen, G.; Abitorabi, A.; Sirenko, O.; Murray, L.; Hill, B. Investigation into an engraftment defect induced by culturing primitive hematopoietic cells with cytokines. Cytotherapy 2001, 3, 307–320. [Google Scholar]
- Ladiges, W.C.; Storb, R.; Thomas, E.D. Canine models of bone marrow transplantation. Lab. Anim. Sci. 1990, 40, 11–15. [Google Scholar]
- Storb, R.; Weiden, P.L.; Graham, T.C.; Thomas, E.D. Studies of marrow transplantation in dogs. Transplant. Proc. 1976, 8, 545–549. [Google Scholar]
- Trobridge, G.D. Genotoxicity of retroviral hematopoietic stem cell gene therapy. Expet. Opin. Biol. Ther. 2011, 11, 581–593. [Google Scholar] [CrossRef]
- Suter, S.E.; Gouthro, T.A.; McSweeney, P.A.; Nash, R.A.; Haskins, M.E.; Felsburg, P.J.; Henthorn, P.S. Isolation and characterization of pediatric canine bone marrow CD34+ cells. Vet. Immunol. Immunopathol. 2004, 101, 31–47. [Google Scholar] [CrossRef]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar]
- Goerner, M.; Bruno, B.; McSweeney, P.A.; Buron, G.; Storb, R.; Kiem, H.P. The use of granulocyte colony-stimulating factor during retroviral transduction on fibronectin fragment CH-296 enhances gene transfer into hematopoietic repopulating cells in dogs. Blood 1999, 94, 2287–2292. [Google Scholar]
- Goerner, M.; Horn, P.A.; Peterson, L.; Kurre, P.; Storb, R.; Rasko, J.E.; Kiem, H.P. Sustained multilineage gene persistence and expression in dogs transplanted with CD34(+) marrow cells transduced by RD114-pseudotype oncoretrovirus vectors. Blood 2001, 98, 2065–2070. [Google Scholar] [CrossRef]
- Lehmann-Che, J.; Renault, N.; Giron, M.L.; Roingeard, P.; Clave, E.; Tobaly-Tapiero, J.; Bittoun, P.; Toubert, A.; de The, H.; Saib, A. Centrosomal latency of incoming foamy viruses in resting cells. PLoS Pathogens 2007, 3, e74. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; Hickstein, D.D. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PloS One 2012, 7, e45173. [Google Scholar]
- Beard, B.C.; Keyser, K.A.; Trobridge, G.D.; Peterson, L.J.; Miller, D.G.; Jacobs, M.; Kaul, R.; Kiem, H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007, 18, 423–434. [Google Scholar] [CrossRef]
- Harrison, D.E. Competitive repopulation: a new assay for long-term stem cell functional capacity. Blood 1980, 55, 77–81. [Google Scholar]
- Kiem, H.P.; Heyward, S.; Winkler, A.; Potter, J.; Allen, J.M.; Miller, A.D.; Andrews, R.G. Gene transfer into marrow repopulating cells: comparison between amphotropic and gibbon ape leukemia virus pseudotyped retroviral vectors in a competitive repopulation assay in baboons. Blood 1997, 90, 4638–4645. [Google Scholar]
- Trobridge, G.D.; Allen, J.; Peterson, L.; Ironside, C.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Hai, M.; Tuschong, L.M.; Burkholder, T.H.; Gu, Y.C.; Sokolic, R.A.; Ferguson, C.; Dunbar, C.E.; Hickstein, D.D. Correction of the disease phenotype in canine leukocyte adhesion deficiency using ex vivo hematopoietic stem cell gene therapy. Blood 2006, 108, 3313–3320. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Olson, E.M.; Huo, Y.; Tuschong, L.M.; Allen, J.M.; Li, Y.; Burkholder, T.H.; Russell, D.W. Treatment of canine leukocyte adhesion deficiency by foamy virus vectors expressing CD18 from a PGK promoter. Gene Ther. 2011, 18, 553–559. [Google Scholar]
- Rivat, C.; Santilli, G.; Gaspar, H.B.; Thrasher, A.J. Gene therapy for primary immunodeficiencies. Hum. Gene Ther. 2012, 23, 668–675. [Google Scholar] [CrossRef]
- Persons, D.A.; Allay, E.R.; Sabatino, D.E.; Kelly, P.; Bodine, D.M.; Nienhuis, A.W. Functional requirements for phenotypic correction of murine beta-thalassemia: implications for human gene therapy. Blood 2001, 97, 3275–3282. [Google Scholar]
- Searcy, G.P.; Miller, D.R.; Tasker, J.B. Congenital hemolytic anemia in the Basenji dog due to erythrocyte pyruvate kinase deficiency. Can. J. Comp. Med. 1971, 35, 67–70. [Google Scholar]
- Whitney, K.M.; Goodman, S.A.; Bailey, E.M.; Lothrop, C.D., Jr. The molecular basis of canine pyruvate kinase deficiency. Exp. Hematol. 1994, 22, 866–874. [Google Scholar]
- Weiden, P.L.; Storb, R.; Graham, T.C.; Schroeder, M.L. Severe hereditary haemolytic anaemica in dogs treated by marrow trasplantation. Br. J. Haematol. 1976, 33, 357–362. [Google Scholar] [CrossRef]
- Zaucha, J.A.; Yu, C.; Lothrop, C.D., Jr.; Nash, R.A.; Sale, G.; Georges, G.; Kiem, H.P.; Niemeyer, G.P.; Dufresne, M.; Cao, Q.; Storb, R. Severe canine hereditary hemolytic anemia treated by nonmyeloablative marrow transplantation. Biol. Blood Marrow Transplant. 2001, 7, 14–24. [Google Scholar]
- Trobridge, G.; Beard, B.C.; Kiem, H.P. Hematopoietic stem cell transduction and amplification in large animal models. Hum. Gene Ther. 2005, 16, 1355–1366. [Google Scholar]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar] [CrossRef]
- Ohmine, K.; Li, Y.; Bauer, T.R., Jr.; Hickstein, D.D.; Russell, D.W. Tracking of specific integrant clones in dogs treated with foamy virus vectors. Hum. Gene Ther. 2011, 22, 217–224. [Google Scholar] [CrossRef]
- Joag, S.V. Primate models of AIDS. Microb. Infect. 2000, 2, 223–229. [Google Scholar]
- Schweizer, M.; Turek, R.; Hahn, H.; Schliephake, A.; Netzer, K.O.; Eder, G.; Reinhardt, M.; Rethwilm, A.; Neumann-Haefelin, D. Markers of foamy virus infections in monkeys, apes, and accidentally infected humans: appropriate testing fails to confirm suspected foamy virus prevalence in human. AIDS Res. Hum. Retroviruses 1995, 11, 161–170. [Google Scholar]
- Trobridge, G.D.; Wu, R.A.; Beard, B.C.; Chiu, S.Y.; Munoz, N.M.; von Laer, D.; Rossi, J.J.; Kiem, H.P. Protection of stem cell-derived lymphocytes in a primate AIDS gene therapy model after in vivo selection. PloS One 2009, 4, e7693. [Google Scholar]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Invest. 2010, 120, 2345–2354. [Google Scholar]
- Horn, P.A.; Tani, K.; Martin, U.; Niemann, H. Nonhuman primates: Embryonic stem cells and transgenesis. Clon. Stem Cell. 2006, 8, 124–129. [Google Scholar] [CrossRef]
- Sasaki, E.; Suemizu, H.; Shimada, A.; Hanazawa, K.; Oiwa, R.; Kamioka, M.; Tomioka, I.; Sotomaru, Y.; Hirakawa, R.; Eto, T.; Shiozawa, S.; Maeda, T.; Ito, M.; Ito, R.; Kito, C.; Yagihashi, C.; Kawai, K.; Miyoshi, H.; Tanioka, Y.; Tamaoki, N.; Habu, S.; Okano, H.; Nomura, T. Generation of transgenic non-human primates with germline transmission. Nature 2009, 459, 523–527. [Google Scholar]
- Wurm, M.; Schambach, A.; Lindemann, D.; Hanenberg, H.; Standker, L.; Forssmann, W.G.; Blasczyk, R.; Horn, P.A. The influence of semen-derived enhancer of virus infection on the efficiency of retroviral gene transfer. J. Gene Med. 2010, 12, 137–146. [Google Scholar]
- Vassilopoulos, G.; Josephson, N.C.; Trobridge, G. Development of foamy virus vectors. Meth. Mol. Med. 2003, 76, 545–564. [Google Scholar]
- Donahue, R.E.; Sorrentino, B.P.; Hawley, R.G.; An, D.S.; Chen, I.S.; Wersto, R.P. Fibronectin fragment CH-296 inhibits apoptosis and enhances ex vivo gene transfer by murine retrovirus and human lentivirus vectors independent of viral tropism in nonhuman primate CD34+ cells. Mol. Ther. 2001, 3, 359–367. [Google Scholar] [CrossRef]
- Horn, P.A.; Wurm, M.; Tani, K.; Lindemann, D.; Sasaki, E.; Blasczyk, R.; Hanenberg, H. Foamyvirus Vectors Efficiently Transduce Nonhuman Primate Hematopoietic and Embryonic Stem Cells. Tissue Antigens 2006, 67, 452. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trobridge, G.D.; Horn, P.A.; Beard, B.C.; Kiem, H.-P. Large Animal Models for Foamy Virus Vector Gene Therapy. Viruses 2012, 4, 3572-3588. https://doi.org/10.3390/v4123572
Trobridge GD, Horn PA, Beard BC, Kiem H-P. Large Animal Models for Foamy Virus Vector Gene Therapy. Viruses. 2012; 4(12):3572-3588. https://doi.org/10.3390/v4123572
Chicago/Turabian StyleTrobridge, Grant D., Peter A. Horn, Brian C. Beard, and Hans-Peter Kiem. 2012. "Large Animal Models for Foamy Virus Vector Gene Therapy" Viruses 4, no. 12: 3572-3588. https://doi.org/10.3390/v4123572
APA StyleTrobridge, G. D., Horn, P. A., Beard, B. C., & Kiem, H. -P. (2012). Large Animal Models for Foamy Virus Vector Gene Therapy. Viruses, 4(12), 3572-3588. https://doi.org/10.3390/v4123572