Diversity in Glycosaminoglycan Binding Amongst hMPV G Protein Lineages

Abstract
:1. Introduction
2. Results and Discussion
2.1. Susceptibility of hMPV Primary Isolates to Inhibition of Infection by Heparin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Subtype | Ct value 2 | |
|---|---|---|---|
| Heparin Pre-treated | Untreated virus | ||
| V47041 | B2 | Negative | 24.1 |
| V52283 | A2 | 30.2 | 16.6 |
| V50569 | B1 | Negative | 13.1 |
2.2. GAG-Deficient CHO Cells Are Resistant to hMPV Infection

2.3. Binding of hMPV G Glycoprotein to Immobilised Heparin
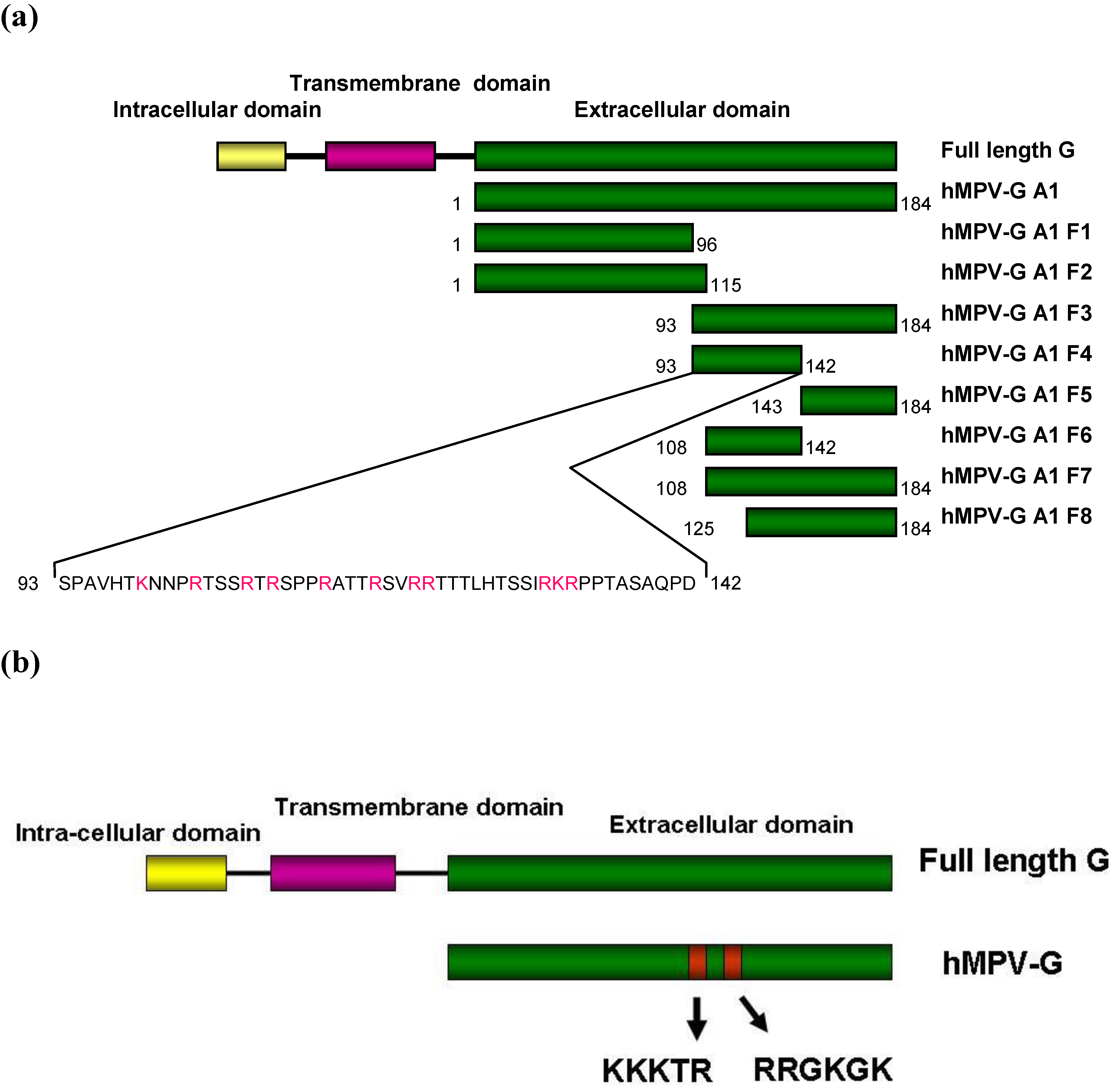
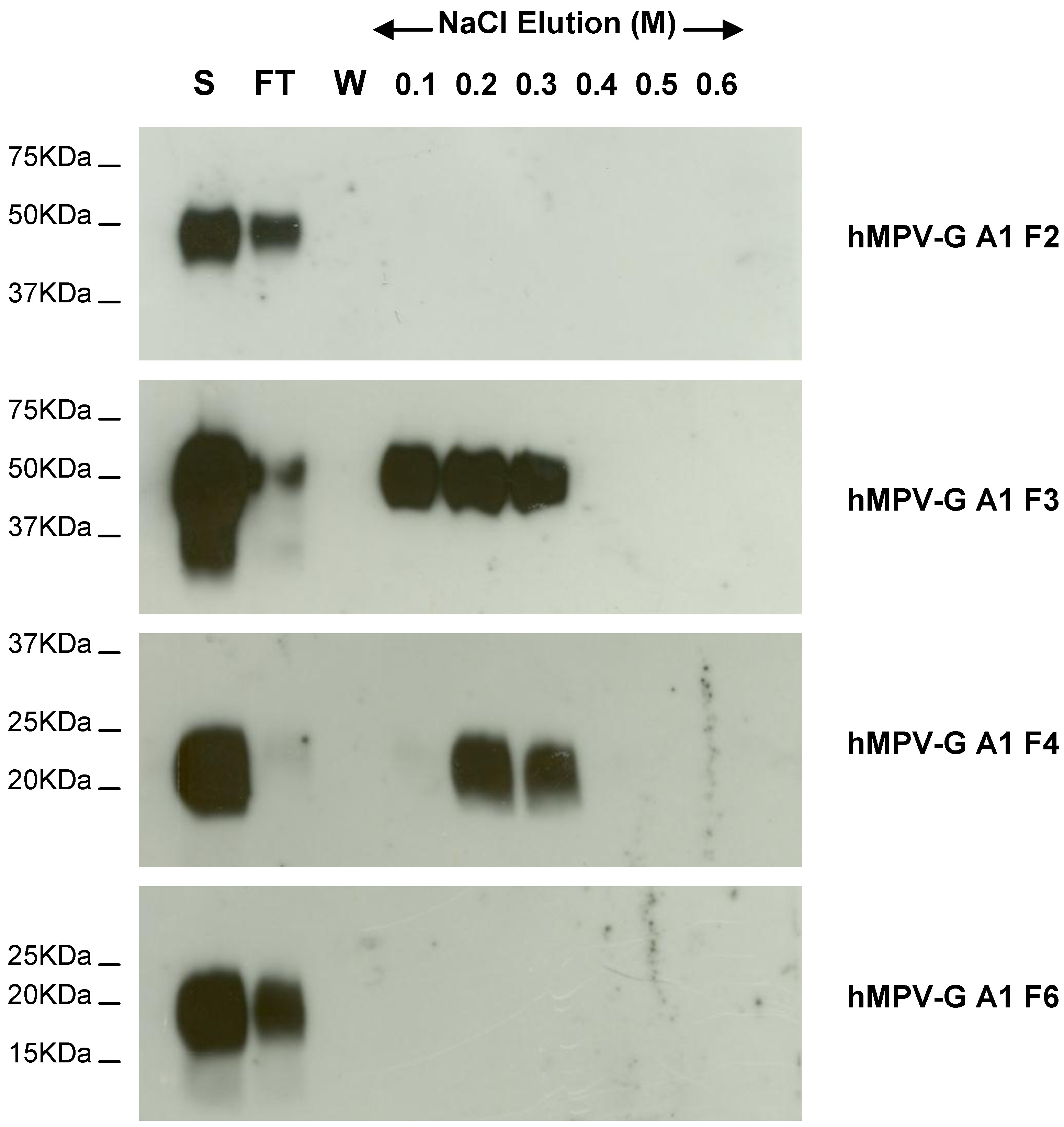
2.4. Functional Domains in hMPV G A1 Protein Involved in GAG Interactions




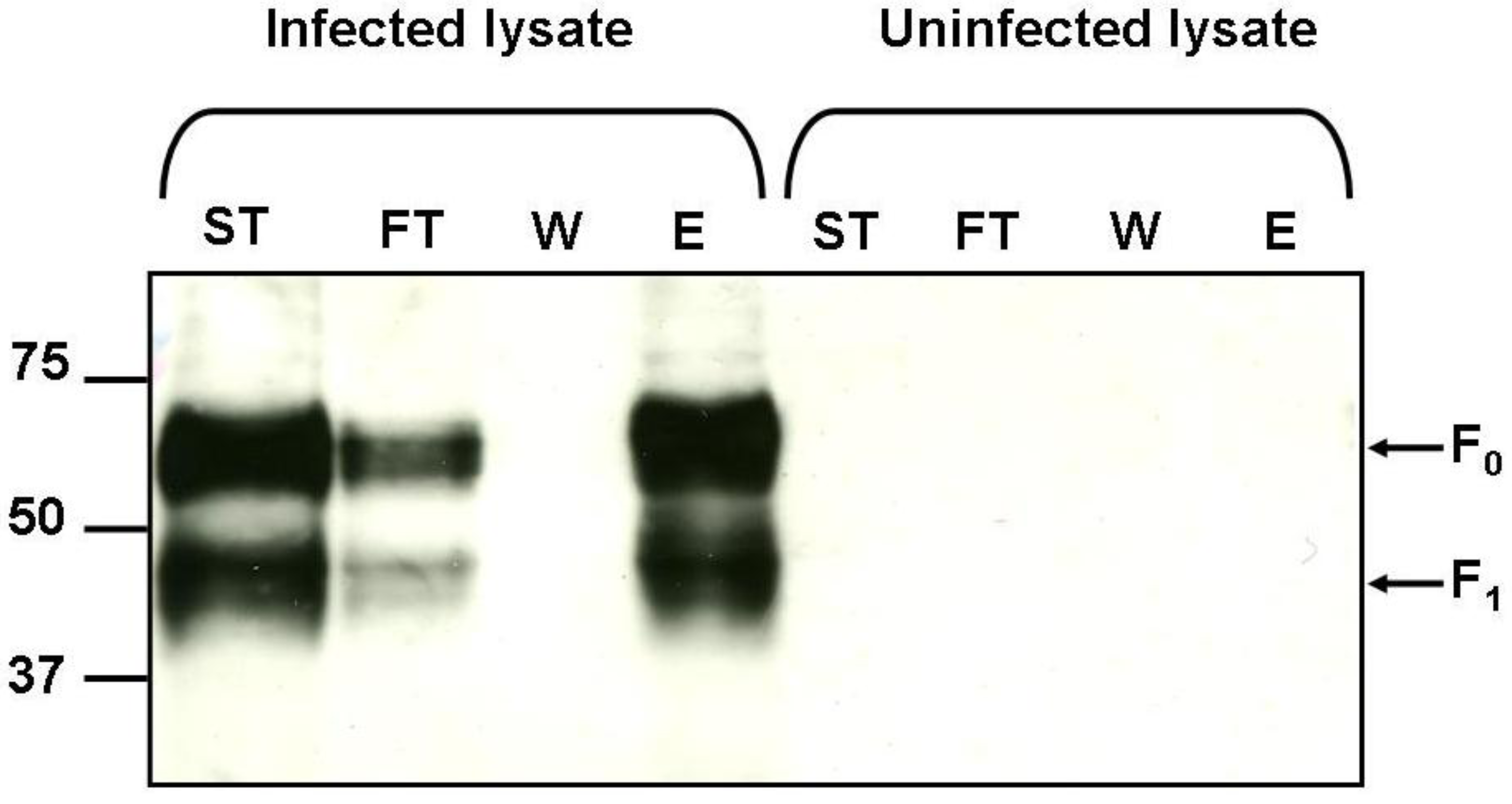
2.5. Binding of hMPV F Protein to Immobilised Heparin


3. Experimental Section
3.1. Cells and Viruses
3.2. Heparin Inhibition of hMPV Infection
3.3. GAG Dependence of hMPV Primary Isolates
3.4. Construction and Expression of Recombinant hMPV G Proteins
3.5. Heparin Agarose Affinity Chromatography
| Primer Name | hMPV Strains | Primer Sequence |
|---|---|---|
| GB2F | B2 | 5'-GGGGAATTCGATCATGCAACATTAAGAAACATG-3' |
| GB2R | B2 | 5'-GGGTCTAGAGCTCCTGCACCTCYCCGTGCAT-3' |
| GB1F | B1 | 5'-AGAATTCGAATCAGAACATCACACCAG-3' |
| GB1R | B1 | 5'-ATCTAGAGCTGTGCTTGCAGATGCCTG-3' |
| GA2F | A2 | 5'-AGAATTCGATTATGCAACATTAAAAAACATG-3' |
| GA2R | A2 | 5'-ATCTAGAGCACTACTTAGAGAAGATGTGTC-3' |
| GA1F | A1 | 5'-AGAATTCAACTATAMAATGCAARAAAACTCCGA-3' |
| GA1R | A1 | 5'-TTCTAGAGCACTAGTTTGGTTGTATGTTGTTGA-3' |
| GA1F2 | A1 | 5'-AGAATTCAGCCCAGCAGTCCACACAAAAAAC-3' |
| GA1R1 | A1 | 5'-TTCTAGATCGACTGCTGGGCTTGTCTTTGTTC-3' |
| GA1R2 | A1 | 5'-TTCTAGATCTGTTGTTGCCCGTGGTGGGGAAC-3' |
| GA1F3 | A1 | 5'-AGAATTCGACAGCAGCGCAACAATCC-3' |
| GA1F4 | A1 | 5'-AGAATTCACACGTTCCCCACCACG-3' |
| GA1F5 | A1 | 5'-AGAATTCCTCCACACAAGCAGCATAAG-3' |
| GA1R4 | A1 | 5'-TTCTAGAGCGTCTGGTTGGGCTGATGC-3' |
| FMPV-for | B2 | 5'-GGGACGCGTCTTAAAGAGAGCTAYYTAGAAG-3' |
| FMPV-r2B | B2 | 5'-GGGTCTAGAGCRCCAGTGTTTCCTTTTTCTGC-3' |
3.6. Detection of hMPV G Protein by Western Blot
3.7. Heparin Binding Studies Using hMPV Infected Cell Lysates
3.8. Detection of hMPV F Protein by Western Blot
3.9. Construction and Expression of hMPV F Protein
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Boivin, G.; Abed, Y.; Pelletier, G.; Ruel, L.; Moisan, D.; Cote, S.; Peret, T.C.; Erdman, D.D.; Anderson, L.J. Virological features and clinical manifestations associated with human metapneumovirus: A new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 2002, 186, 1330–1334. [Google Scholar] [CrossRef]
- Boivin, G.; de Serres, G.; Cote, S.; Gilca, R.; Abed, Y.; Rochette, L.; Bergeron, M.G.; Dery, P. Human metapneumovirus infections in hospitalized children. Emerg. Infect. Dis. 2003, 9, 634–640. [Google Scholar] [CrossRef]
- Osterhaus, A.; Fouchier, R. Human metapneumovirus in the community. Lancet 2003, 361, 890–891. [Google Scholar] [CrossRef]
- Peret, T.C.; Boivin, G.; Li, Y.; Couillard, M.; Humphrey, C.; Osterhaus, A.D.; Erdman, D.D.; Anderson, L.J. Characterization of human metapneumoviruses isolated from patients in North America. J. Infect. Dis. 2002, 185, 1660–1663. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef]
- Kahn, J.S. Epidemiology of human metapneumovirus. Clin. Microbiol. Rev. 2006, 19, 546–557. [Google Scholar] [CrossRef]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus taxonomy: Ninth report of the international committee on taxonomy of viruses; Ninth Report. Academic Press: Elsevier; Oxford, UK, 2012; pp. 672–682. Available online: http://ictvonline.org/virusTaxonomy.asp?version=2009 (accessed on 14 February 2012).
- Van den Hoogen, B.G.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Analysis of the genomic sequence of a human metapneumovirus. Virology 2002, 295, 119–132. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; van Doornum, G.J.; Fockens, J.C.; Cornelissen, J.J.; Beyer, W.E.; de Groot, R.; Osterhaus, A.D.; Fouchier, R.A. Prevalence and clinical symptoms of human metapneumovirus infection in hospitalized patients. J. Infect. Dis. 2003, 188, 1571–1577. [Google Scholar] [CrossRef]
- Cilla, G.; Onate, E.; Perez-Yarza, E.G.; Montes, M.; Vicente, D.; Perez-Trallero, E. Hospitalization rates for human metapneumovirus infection among 0- to 3-year-olds in Gipuzkoa (Basque Country), Spain. Epidemiol. Infect. 2009, 137, 66–72. [Google Scholar] [CrossRef]
- Ordas, J.; Boga, J.A.; Alvarez-Arguelles, M.; Villa, L.; Rodriguez-Dehli, C.; de Ona, M.; Rodriguez, J.; Melon, S. Role of metapneumovirus in viral respiratory infections in young children. J. Clin. Microbiol. 2006, 44, 2739–2742. [Google Scholar] [CrossRef]
- Bastien, N.; Liu, L.; Ward, D.; Taylor, T.; Li, Y. Genetic variability of the G glycoprotein gene of human metapneumovirus. J. Clin. Microbiol. 2004, 42, 3532–3537. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Boivin, G.; Hanson, C.T.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Genetic diversity between human metapneumovirus subgroups. Virology 2003, 315, 1–9. [Google Scholar] [CrossRef]
- Ishiguro, N.; Ebihara, T.; Endo, R.; Ma, X.; Kikuta, H.; Ishiko, H.; Kobayashi, K. High genetic diversity of the attachment (G) protein of human metapneumovirus. J. Clin. Microbiol. 2004, 42, 3406–3414. [Google Scholar] [CrossRef]
- Ludewick, H.P.; Abed, Y.; van Niekerk, N.; Boivin, G.; Klugman, K.P.; Madhi, S.A. Human metapneumovirus genetic variability, South Africa. Emerg. Infect. Dis. 2005, 11, 1074–1078. [Google Scholar]
- van den Hoogen, B.G.; Herfst, S.; Sprong, L.; Cane, P.A.; Forleo-Neto, E.; de Swart, R.L.; Osterhaus, A.D.; Fouchier, R.A. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 2004, 10, 658–666. [Google Scholar]
- Feuillet, F.; Lina, B.; Rosa-Calatrava, M.; Boivin, G. Ten years of human metapneumovirus research. J. Clin. Virol. 2012, 53, 97–105. [Google Scholar] [CrossRef]
- Chanock, R.M.; Murphy, B.R.; Collins, P.L. Parainfluenza viruses. In Fields Virology, 4th; Knipe, D.M., Howley, P.M., Griffin, D.E., Martin, M.A., Lamb, R.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2001; Volume 1, pp. 1341–1379. [Google Scholar]
- Cseke, G.; Maginnis, M.S.; Cox, R.G.; Tollefson, S.J.; Podsiad, A.B.; Wright, D.W.; Dermody, T.S.; Williams, J.V. Integrin αvβ1 promotes infection by human metapneumovirus. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1566–1571. [Google Scholar]
- Skiadopoulos, M.H.; Biacchesi, S.; Buchholz, U.J.; Amaro-Carambot, E.; Surman, S.R.; Collins, P.L.; Murphy, B.R. Individual contributions of the human metapneumovirus F, G, and SH surface glycoproteins to the induction of neutralizing antibodies and protective immunity. Virology 2006, 345, 492–501. [Google Scholar] [CrossRef]
- Skiadopoulos, M.H.; Biacchesi, S.; Buchholz, U.J.; Riggs, J.M.; Surman, S.R.; Amaro-Carambot, E.; McAuliffe, J.M.; Elkins, W.R.; St Claire, M.; Collins, P.L.; et al. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J. Virol. 2004, 78, 6927–6937. [Google Scholar]
- Tang, R.S.; Mahmood, K.; Macphail, M.; Guzzetta, J.M.; Haller, A.A.; Liu, H.; Kaur, J.; Lawlor, H.A.; Stillman, E.A.; Schickli, J.H.; et al. A host-range restricted parainfluenza virus type 3 (PIV3) expressing the human metapneumovirus (hMPV) fusion protein elicits protective immunity in African green monkeys. Vaccine 2005, 23, 1657–1667. [Google Scholar] [CrossRef]
- Liu, L.; Bastien, N.; Li, Y. Intracellular processing, glycosylation, and cell surface expression of human metapneumovirus attachment glycoprotein. J. Virol. 2007, 81, 13435–13443. [Google Scholar] [CrossRef]
- Thammawat, S.; Sadlon, T.A.; Hallsworth, P.G.; Gordon, D.L. Role of cellular glycosaminoglycans and charged regions of viral G protein in human metapneumovirus infection. J. Virol. 2008, 82, 11767–11774. [Google Scholar] [CrossRef]
- Biacchesi, S.; Pham, Q.N.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2–2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 2005, 79, 12608–12613. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and sulfated glycosaminoglycans. In Essentials of Glycobiology, 2nd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 16. [Google Scholar]
- Baba, M.; Snoeck, R.; Pauwels, R.; de Clercq, E. Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, and human immunodeficiency virus. Antimicrob. Agents Chemother. 1988, 32, 1742–1745. [Google Scholar] [CrossRef]
- Feldman, S.A.; Hendry, R.M.; Beeler, J.A. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 1999, 73, 6610–6617. [Google Scholar]
- Hosoya, M.; Balzarini, J.; Shigeta, S.; de Clercq, E. Differential inhibitory effects of sulfated polysaccharides and polymers on the replication of various myxoviruses and retroviruses, depending on the composition of the target amino acid sequences of the viral envelope glycoproteins. Antimicrob. Agents Chemother. 1991, 35, 2515–2520. [Google Scholar] [CrossRef]
- Zhu, Z.; Gershon, M.D.; Ambron, R.; Gabel, C.; Gershon, A.A. Infection of cells by varicella zoster virus: Inhibition of viral entry by mannose 6-phosphate and heparin. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3546–3550. [Google Scholar] [CrossRef]
- Krusat, T.; Streckert, H.J. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 1997, 142, 1247–1254. [Google Scholar] [CrossRef]
- Wyde, P.R.; Moylett, E.H.; Chetty, S.N.; Jewell, A.; Bowlin, T.L.; Piedra, P.A. Comparison of the inhibition of human metapneumovirus and respiratory syncytial virus by NMSO3 in tissue culture assays. Antivir. Res. 2004, 63, 51–59. [Google Scholar]
- Esko, J.D.; Stewart, T.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 3197–3201. [Google Scholar] [CrossRef]
- Maertzdorf, J.; Wang, C.K.; Brown, J.B.; Quinto, J.D.; Chu, M.; de Graaf, M.; van den Hoogen, B.G.; Spaete, R.; Osterhaus, A.D.; Fouchier, R.A. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J. Clin. Microbiol. 2004, 42, 981–986. [Google Scholar] [CrossRef]
- Shenoy-Scaria, A.M.; Gauen, L.K.; Kwong, J.; Shaw, A.S.; Lublin, D.M. Palmitylation of an amino-terminal cysteine motif of protein tyrosine kinases p56lck and p59fyn mediates interaction with glycosyl-phosphatidylinositol-anchored proteins. Mol. Cell Biol. 1993, 13, 6385–6392. [Google Scholar]
- Byrnes, A.P.; Griffin, D.E. Binding of sindbis virus to cell surface heparan sulfate. J. Virol. 1998, 72, 7349–7356. [Google Scholar]
- Heil, M.L.; Albee, A.; Strauss, J.H.; Kuhn, R.J. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 2001, 75, 6303–6309. [Google Scholar] [CrossRef]
- Klimstra, W.B.; Ryman, K.D.; Johnston, R.E. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 1998, 72, 7357–7366. [Google Scholar]
- Garcia-Beato, R.; Martinez, I.; Franci, C.; Real, F.X.; Garcia-Barreno, B.; Melero, J.A. Host cell effect upon glycosylation and antigenicity of human respiratory syncytial virus G glycoprotein. Virology 1996, 221, 301–309. [Google Scholar] [CrossRef]
- Cregg, J.M.; Vedvick, T.S.; Raschke, W.C. Recent advances in the expression of foreign genes in Pichia pastoris. Biotechnology 1993, 11, 905–910. [Google Scholar]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein (KCP) binds to heparin and cell surfaces via positively charged amino acids in CCP1–2. Mol. Immunol. 2006, 43, 1665–1675. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Margalit, H.; Fischer, N.; Ben-Sasson, S.A. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem. 1993, 268, 19228–19231. [Google Scholar]
- Germi, R.; Crance, J.M.; Garin, D.; Guimet, J.; Lortat-Jacob, H.; Ruigrok, R.W.; Zarski, J.P.; Drouet, E. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 2002, 292, 162–168. [Google Scholar] [CrossRef]
- Hallak, L.K.; Spillmann, D.; Collins, P.L.; Peeples, M.E. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J. Virol. 2000, 74, 10508–10513. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Yang, L.; Lamirande, E.W.; Tran, K.C.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: Deletion of G yields a promising vaccine candidate. J. Virol. 2004, 78, 12877–12887. [Google Scholar]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Clements-Mann, M.L.; Harris, D.O.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 13961–13966. [Google Scholar]
- Teng, M.N.; Whitehead, S.S.; Collins, P.L. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology 2001, 289, 283–296. [Google Scholar] [CrossRef]
- Haywood, A.M. Virus receptors: Binding, adhesion strengthening, and changes in viral structure. J. Virol. 1994, 68, 1–5. [Google Scholar]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αυβ3 and αυβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Fuller, A.O.; Lee, W.C. Herpes simplex virus type 1 entry through a cascade of virus-cell interactions requires different roles of gD and gH in penetration. J. Virol. 1992, 66, 5002–5012. [Google Scholar]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar]
- Johnson, R.M.; Spear, P.G. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 1989, 63, 819–827. [Google Scholar]
- McClain, D.S.; Fuller, A.O. Cell-specific kinetics and efficiency of herpes simplex virus type 1 entry are determined by two distinct phases of attachment. Virology 1994, 198, 690–702. [Google Scholar] [CrossRef]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar]
- Chang, A.; Masante, C.; Buchholz, U.J.; Dutch, R.E. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulphate. J. Virol. 2012, 86, 3230–3243. [Google Scholar] [CrossRef]
- Cox, R.G.; Livesay, S.B.; Johnson, M.; Ohi, M.D; Williams, J.V. The human metapneumovirus fusion protein mediates entry via an interaction with RGD-binding integrins. J. Virol. 2012, 86, 12148–12160. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adamson, P.; Thammawat, S.; Muchondo, G.; Sadlon, T.; Gordon, D. Diversity in Glycosaminoglycan Binding Amongst hMPV G Protein Lineages. Viruses 2012, 4, 3785-3803. https://doi.org/10.3390/v4123785
Adamson P, Thammawat S, Muchondo G, Sadlon T, Gordon D. Diversity in Glycosaminoglycan Binding Amongst hMPV G Protein Lineages. Viruses. 2012; 4(12):3785-3803. https://doi.org/10.3390/v4123785
Chicago/Turabian StyleAdamson, Penelope, Sutthiwan Thammawat, Gamaliel Muchondo, Tania Sadlon, and David Gordon. 2012. "Diversity in Glycosaminoglycan Binding Amongst hMPV G Protein Lineages" Viruses 4, no. 12: 3785-3803. https://doi.org/10.3390/v4123785
APA StyleAdamson, P., Thammawat, S., Muchondo, G., Sadlon, T., & Gordon, D. (2012). Diversity in Glycosaminoglycan Binding Amongst hMPV G Protein Lineages. Viruses, 4(12), 3785-3803. https://doi.org/10.3390/v4123785