Immune Responses to West Nile Virus Infection in the Central Nervous System

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virology and Pathogenesis
3. WNV-Induced Pathology in the CNS
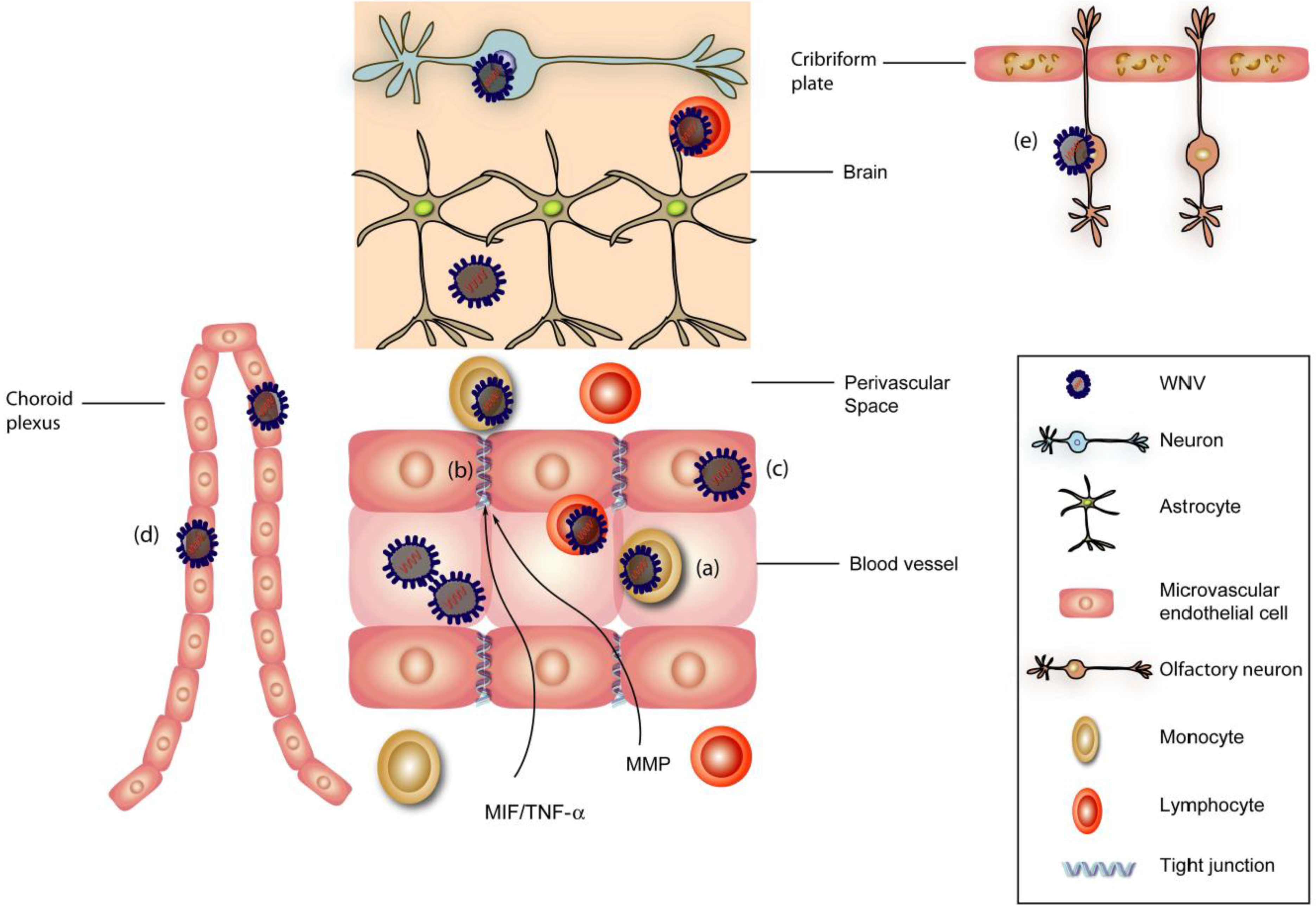
4. Neuroinvasion

5. Neuronal Injury
6. CNS Immune Responses to WNV
6.1. CNS Innate Immunity
6.2. Inflammatory Responses
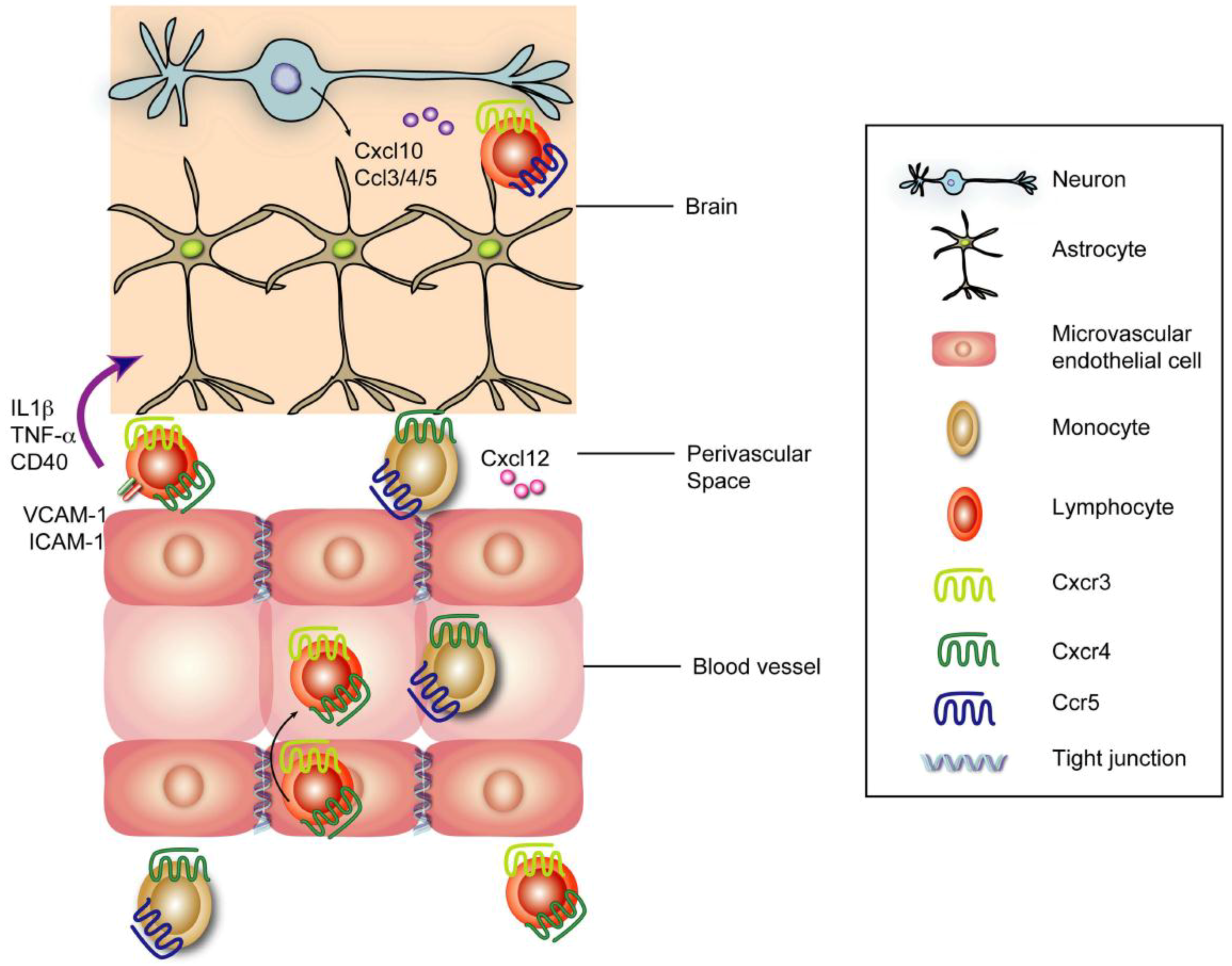
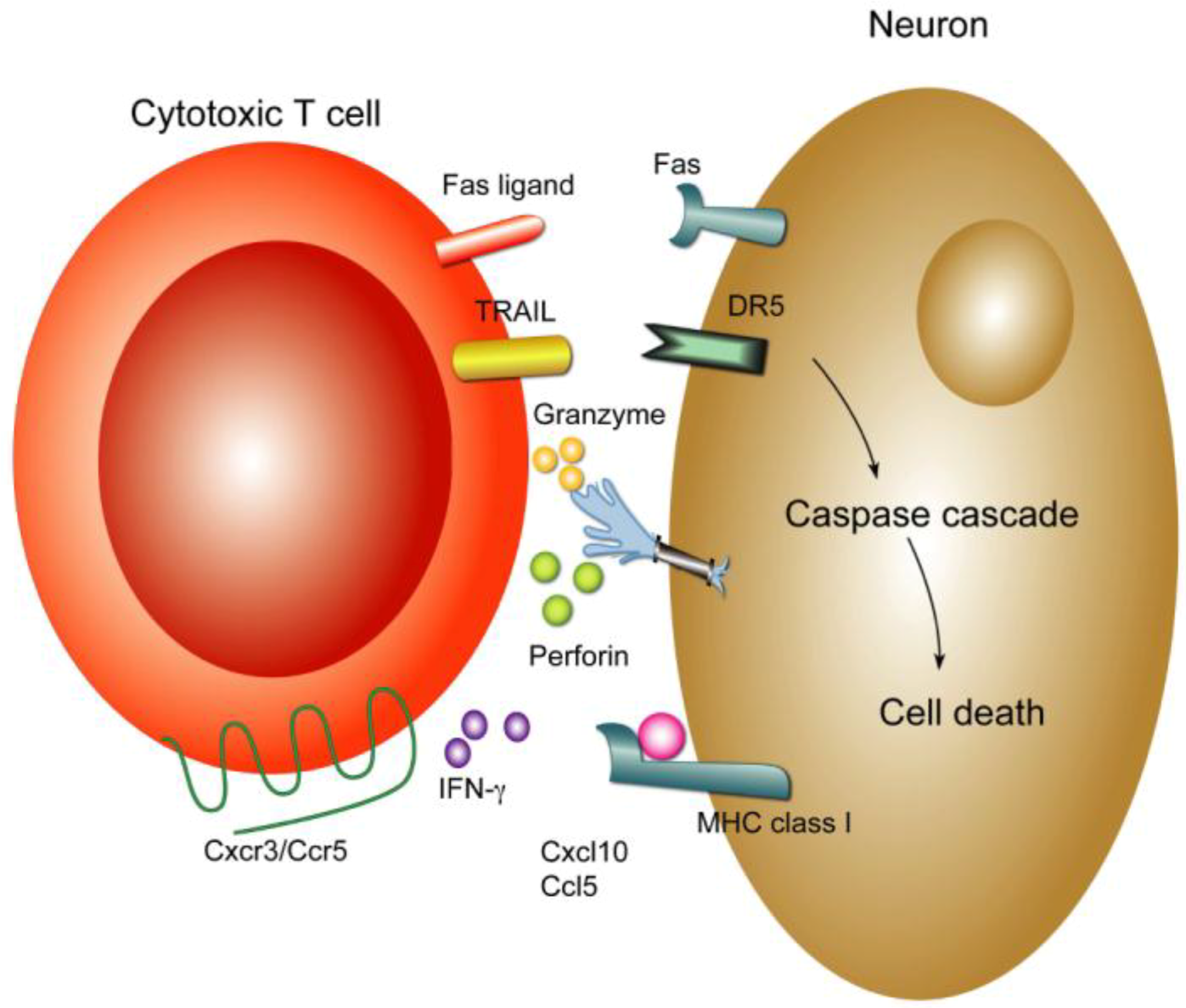
6.3. Cellular Immunity


7. Viral Persistence in the CNS
8. Summary and Future Perspectives
Conflict of Interest
Acknowledgements
References
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and transmission dynamics of West Nile virus disease. Emerging Infect. Dis. 2005, 11, 1167–1173. [Google Scholar] [CrossRef]
- Smithburn, K.C.; Hughes, T.P.; Burke, A.W.; Paul, J.H. A Neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop Med. Hyg. 1940, 1, 471–492. [Google Scholar]
- Kleiboeker, S.B. West Nile Virus. In Encyclopedia of Environmental Health; Nriagu, J.O., Ed.; Elsevier: Burlington, Canada, 2011; pp. 761–768. [Google Scholar]
- Hubálek, Z.; Halouzka, J. West nile fever—A reemerging mosquito-borne viral disease in Europe. Emerging Infect. Dis. 1999, 5, 643–650. [Google Scholar] [CrossRef]
- Omalu, B.I.; Shakir, A.A.; Wang, G.; Lipkin, W.I.; Wiley, C.A. Fatal fulminant pan-meningo-polioencephalitis due to West Nile virus. Brain Pathol. 2003, 13, 465–472. [Google Scholar]
- Armah, H.B.; Wang, G.; Omalu, B.I.; Tesh, R.B.; Gyure, K.A.; Chute, D.J.; Smith, R.D.; Dulai, P.; Vinters, H.V.; Kleinschmidt-DeMasters, B.K.; Wiley, C.A. Systemic distribution of West Nile virus infection: Postmortem immunohistochemical study of six cases. Brain Pathol. 2007, 17, 354–362. [Google Scholar] [CrossRef]
- Busch, M.P.; Wright, D.J.; Custer, B.; Tobler, L.H.; Stramer, S.L.; Kleinman, S.H.; Prince, H.E.; Bianco, C.; Foster, G.; Petersen, L.R.; et al. West nile virus infections projected from blood donor screening data, United States, 2003. Emerg. Infect. Dis. 2006, 12, 395–402. [Google Scholar]
- Leis, A.A.; Fratkin, J.; Stokic, D.S.; Harrington, T.; Webb, R.M.; Slavinski, S.A. West Nile poliomyelitis. Lancet Infect. Dis. 2003, 3, 9–10. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Haddad, M.B.; Tierney, B.C.; Campbell, G.L.; Marfin, A.A.; Van Gerpen, J.A.; Fleischauer, A.; Leis, A.A.; Stokic, D.S.; Petersen, L.R. Neurologic manifestations and outcome of West Nile virus infection. JAMA 2003, 290, 511–515. [Google Scholar]
- Sejvar, J.J.; Marfin, A.A. Manifestations of West Nile neuroinvasive disease. Rev. Med. Virol. 2006, 16, 209–224. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Bode, A.V.; Marfin, A.A.; Campbell, G.L.; Pape, J.; Biggerstaff, B.J.; Petersen, L.R. West Nile Virus-associated flaccid paralysis outcome. Emerging Infect. Dis. 2006, 12, 514–516. [Google Scholar] [CrossRef]
- Sejvar, J.J. The long-term outcomes of human West Nile virus infection. Clin. Infect. Dis. 2007, 44, 1617–1624. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Davis, L.E.; Szabados, E.; Jackson, A.C. Delayed-onset and recurrent limb weakness associated with West Nile virus infection. J. Neurovirol. 2010, 16, 93–100. [Google Scholar] [CrossRef]
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O’Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M.; et al. The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar]
- Chowers, M.Y.; Lang, R.; Nassar, F.; Ben-David, D.; Giladi, M.; Rubinshtein, E.; Itzhaki, A.; Mishal, J.; Siegman-Igra, Y.; Kitzes, R.; et al. Clinical characteristics of the West Nile fever outbreak, Israel, 2000. Emerging Infect. Dis. 2001, 7, 675–678. [Google Scholar]
- Huhn, G.D.; Austin, C.; Langkop, C.; Kelly, K.; Lucht, R.; Lampman, R.; Novak, R.; Haramis, L.; Boker, R.; Smith, S.; et al. The emergence of west nile virus during a large outbreak in Illinois in 2002. Am. J. Trop. Med. Hyg. 2005, 72, 768–776. [Google Scholar]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef]
- Mashimo, T.; Lucas, M.; Simon-Chazottes, D.; Frenkiel, M.-P.; Montagutelli, X.; Ceccaldi, P.-E.; Deubel, V.; Guenet, J.-L.; Despres, P. A nonsense mutation in the gene encoding 2’-5’-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11311–11316. [Google Scholar]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Bigham, A.W.; Buckingham, K.J.; Husain, S.; Emond, M.J.; Bofferding, K.M.; Gildersleeve, H.; Rutherford, A.; Astakhova, N.M.; Perelygin, A.A.; Busch, M.P.; et al. Host genetic risk factors for West Nile Virus infection and disease progression. PLoS ONE 2011, 6, e24745. [Google Scholar]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Brinton, M.A. The molecular biology of West Nile Virus: A new invader of the western hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef]
- Guirakhoo, F.; Bolin, R.A.; Roehrig, J.T. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology 1992, 191, 921–931. [Google Scholar] [CrossRef]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar]
- Mukhopadhyay, S.; Kim, B.-S.; Chipman, P.R.; Rossmann, M.G.; Kuhn, R.J. Structure of West Nile virus. Science 2003, 302, 248. [Google Scholar]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Invest. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef]
- Lim, P.-Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.-Y.; Bernard, K.A. Keratinocytes are cell targets of West Nile Virus in vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar] [CrossRef]
- Bourne, N.; Scholle, F.; Silva, M.C.; Rossi, S.L.; Dewsbury, N.; Judy, B.; De Aguiar, J.B.; Leon, M.A.; Estes, D.M.; Fayzulin, R.; Mason, P.W. Early production of type I interferon during West Nile virus infection: Role for lymphoid tissues in IRF3-independent interferon production. J. Virol. 2007, 81, 9100–9108. [Google Scholar]
- Purtha, W.E.; Chachu, K.A.; Virgin, H.W., 4th; Diamond, M.S. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J. Virol. 2008, 82, 10964–10974. [Google Scholar] [CrossRef]
- Wang, T.; Scully, E.; Yin, Z.; Kim, J.H.; Wang, S.; Yan, J.; Mamula, M.; Anderson, J.F.; Craft, J.; Fikrig, E. IFN-γ-Producing γδ T cells help control murine West Nile Virus infection. J. Immunol. 2003, 171, 2524–2531. [Google Scholar]
- Vargin, V.V.; Semenov, B.F. Changes of natural killer cell activity in different mouse lines by acute and asymptomatic flavivirus infections. Acta. Virol. 1986, 30, 303–308. [Google Scholar]
- Bai, F.; Kong, K.-F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Eldadah, A.H.; Nathanson, N. Pathogenesis of West Nile Virus encepahlitis in mice and rats. II. Virus multiplication, evolution of immunofluorescence, and development of histological lesions in the brain. Am. J. Epidemiol. 1967, 86, 776–790. [Google Scholar]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Shrestha, B.; Samuel, M.A.; Diamond, M.S. CD8+ T cells require perforin to clear West Nile Virus from infected neurons. J. Virol. 2006, 80, 119–129. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of west Nile Virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Shrestha, B.; Pinto, A.K.; Green, S.; Bosch, I.; Diamond, M.S. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J. Virol. 2012, 86, 8937–8948. [Google Scholar] [CrossRef]
- Daffis, S.; Samuel, M.A.; Keller, B.C.; Gale, M.; Diamond, M.S. Cell-Specific IRF-3 responses protect against west Nile Virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007, 3, e106. [Google Scholar] [CrossRef]
- Chambers, T.J.; Diamond, M.S. Pathogenesis of flavivirus encephalitis. Adv. Virus Res. 2003, 60, 273–342. [Google Scholar] [CrossRef]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Diamond, M.S.; Klein, R.S. West Nile virus: Crossing the blood-brain barrier. Nat. Med. 2004, 10, 1294–1295. [Google Scholar] [CrossRef]
- Diniz, J.A.P.; Da Rosa, A.P.A.T.; Guzman, H.; Xu, F.; Xiao, S.-Y.; Popov, V.L.; Vasconcelos, P.F.C.; Tesh, R.B. West Nile virus infection of primary mouse neuronal and neuroglial cells: The role of astrocytes in chronic infection. Am. J. Trop. Med. Hyg. 2006, 75, 691–696. [Google Scholar]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M., Jr; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003, 77, 13203–13213. [Google Scholar] [CrossRef]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar]
- Morrey, J.D.; Siddharthan, V.; Wang, H.; Hall, J.O.; Skirpstunas, R.T.; Olsen, A.L.; Nordstrom, J.L.; Koenig, S.; Johnson, S.; Diamond, M.S. West Nile virus–induced acute flaccid paralysis is prevented by monoclonal antibody treatment when administered after infection of spinal cord neurons. J. Neurovirol. 2008, 14, 152–163. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Johnson, R.T.; Mims, C.A. Pathogenesis of Viral infections of the nervous system. New Engl. J. Med. 1968, 278, 84–92. [Google Scholar] [CrossRef]
- Johnson, R.T.; Mims, C.A. Pathogenesis of viral infections of the nervous system. New Engl. J. Med. 1968, 278, 23–30. [Google Scholar] [CrossRef]
- de Vries, H.E.; Blom-Roosemalen, M.C.M.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J. C.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Fiala, M.; Looney, D.J.; Stins, M.; Way, D.D.; Zhang, L.; Gan, X.; Chiappelli, F.; Schweitzer, E.S.; Shapshak, P.; Weinand, M.; et al. TNF-alpha opens a paracellular route for HIV-1 invasion across the blood-brain barrier. Mol. Med. 1997, 3, 553–564. [Google Scholar]
- Sultana, H.; Neelakanta, G.; Foellmer, H.G.; Montgomery, R.R.; Anderson, J.F.; Koski, R.A.; Medzhitov, R.M.; Fikrig, E. Semaphorin 7A contributes to West Nile virus pathogenesis through TGF-β1/Smad6 signaling. J. Immunol. 2012, 189, 3150–3158. [Google Scholar] [CrossRef]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.-F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal of West Nile virus-induced blood–brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 2010, 397, 130–138. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. J. Neurovirol. 2006, 12, 129–139. [Google Scholar] [CrossRef]
- Monath, T.P.; Cropp, C.B.; Harrison, A.K. Mode of entry of a neurotropic arbovirus into the central nervous system. Reinvestigation of an old controversy. Lab. Invest. 1983, 48, 399–410. [Google Scholar]
- Dietzschold, B.; Schnell, M.; Koprowski, H. Pathogenesis of rabies. Curr. Top. Microbiol. Immunol. 2005, 292, 45–56. [Google Scholar]
- Kramer-Hämmerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.M.; Wang, T. Drak2 contributes to West Nile virus entry into the brain and lethal encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar]
- Garcia-Tapia, D.; Loiacono, C.M.; Kleiboeker, S.B. Replication of West Nile virus in equine peripheral blood mononuclear cells. Vet. Immunol. Immunopathol. 2006, 110, 229–244. [Google Scholar] [CrossRef]
- Brown, A.N.; Kent, K.A.; Bennett, C.J.; Bernard, K.A. Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 2007, 368, 422. [Google Scholar] [CrossRef]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef]
- Beasley, D.W.C.; Li, L.; Suderman, M.T.; Barrett, A.D.T. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Samuel, M.A.; Morrey, J.D.; Diamond, M.S. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile Virus encephalitis. J. Virol. 2007, 81, 2614–2623. [Google Scholar] [CrossRef]
- Ramanathan, M.P.; Chambers, J.A.; Pankhong, P.; Chattergoon, M.; Attatippaholkun, W.; Dang, K.; Shah, N.; Weiner, D.B. Host cell killing by the West Nile Virus NS2B–NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 2006, 345, 56–72. [Google Scholar] [CrossRef]
- del Carmen Parquet, M.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Letters 2001, 500, 17–24. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; DeFilippis, V.; Früh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile Virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar]
- Hail, N.; Carter, B.; Konopleva, M.; Andreeff, M. Apoptosis effector mechanisms: A requiem performed in different keys. Apoptosis 2006, 11, 889–904. [Google Scholar] [CrossRef]
- Kroemer, G.; Martin, S.J. Caspase-independent cell death. Nat. Med. 2005, 11, 725–730. [Google Scholar] [CrossRef]
- Chu, J.J.H.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 2010, 7, 73. [Google Scholar] [CrossRef]
- Zhang, B.; Patel, J.; Croyle, M.; Diamond, M.S.; Klein, R.S. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J. Neuroimmunol. 2010, 224, 28–38. [Google Scholar] [CrossRef]
- Bradl, M.; Hohlfeld, R. Molecular pathogenesis of neuroinflammation. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1364–1370. [Google Scholar] [CrossRef]
- Power, C.; Johnson, R.T. Neuroimmune and neurovirological aspects of human immunodeficiency virus infection. Adv. Virus Res. 2001, 56, 389–433. [Google Scholar] [CrossRef]
- Chen, C.-J.; Ou, Y.-C.; Lin, S.-Y.; Raung, S.-L.; Liao, S.-L.; Lai, C.-Y.; Chen, S.-Y.; Chen, J.-H. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol. 2010, 91, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Schachtele, S.; Hu, S.; Little, M.; Lokensgard, J. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroimmunol. 2010, 7, 35. [Google Scholar]
- McGavern, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011, 11, 318–329. [Google Scholar] [CrossRef]
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M., Jr; Diamond, M.S. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M., Jr; Diamond, M.S. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 2010, 84, 12125–12138. [Google Scholar]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.T.; Qian, F.; Montgomery, R.R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009, 30, 242–253. [Google Scholar] [CrossRef]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M., Jr; Diamond, M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 2008, 82, 8465–8475. [Google Scholar] [CrossRef]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar]
- Samuel, M.A.; Whitby, K.; Keller, B.C.; Marri, A.; Barchet, W.; Williams, B.R.G.; Silverman, R.H.; Gale, M., Jr; Diamond, M.S. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006, 80, 7009–7019. [Google Scholar]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar]
- Zhang, B.; Chan, Y.K.; Lu, B.; Diamond, M.S.; Klein, R.S. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 2008, 180, 2641–2649. [Google Scholar]
- Glass, W.G.; Lim, J.K.; Cholera, R.; Pletnev, A.G.; Gao, J.-L.; Murphy, P.M. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005, 202, 1087–1098. [Google Scholar] [CrossRef]
- Shirato, K.; Kimura, T.; Mizutani, T.; Kariwa, H.; Takashima, I. Different chemokine expression in lethal and non-lethal murine West Nile virus infection. J. Med. Virol. 2004, 74, 507–513. [Google Scholar] [CrossRef]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Müller, M.; Rana, S.; Shrestha, B.; Radford, J.; Rooijen, N.V.; Campbell, I.L.; King, N.J. C. Ly6c+ “nflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 2008, 205, 2319–2337. [Google Scholar] [CrossRef]
- Lim, J.K.; Obara, C.J.; Rivollier, A.; Pletnev, A.G.; Kelsall, B.L.; Murphy, P.M. Chemokine Receptor Ccr2 is critical for monocyte accumulation and survival in west nile virus encephalitis. J. Immunol. 2011, 186, 471–478. [Google Scholar] [CrossRef]
- Wang, P.; Bai, F.; Zenewicz, L.A.; Dai, J.; Gate, D.; Cheng, G.; Yang, L.; Qian, F.; Yuan, X.; Montgomery, R.R.; Flavell, R.A.; Town, T.; Fikrig, E. IL-22 Signaling contributes to west nile encephalitis pathogenesis. PLoS ONE 2012, 7, e44153. [Google Scholar]
- Sitati, E.M.; Diamond, M.S. CD4+ T-Cell responses are required for clearance of west Nile virus from the central nervous system. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef]
- Sitati, E.; McCandless, E.E.; Klein, R.S.; Diamond, M.S. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile Virus encephalitis. J. Virol. 2007, 81, 9801–9811. [Google Scholar] [CrossRef]
- Shrestha, B.; Zhang, B.; Purtha, W.E.; Klein, R.S.; Diamond, M.S. Tumor necrosis factor alpha protects against lethal West Nile Virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J. Virol. 2008, 82, 8956–8964. [Google Scholar] [CrossRef]
- Chevalier, G.; Suberbielle, E.; Monnet, C.; Duplan, V.; Martin-Blondel, G.; Farrugia, F.; Le Masson, G.; Liblau, R.; Gonzalez-Dunia, D. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog. 2011, 7, e1002393. [Google Scholar] [CrossRef]
- Harty, J.T.; Badovinac, V.P. Influence of effector molecules on the CD8+ T cell response to infection. Curr. Opin. Immunol. 2002, 14, 360–365. [Google Scholar]
- Russell, J.H.; Ley, T.J. Lymphocyte-Mediated Cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef]
- Shresta, S.; Pham, C.T.; Thomas, D.A.; Graubert, T.A.; Ley, T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998, 10, 581–587. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile Virus infection in the central nervous system. J. Virol. 2007, 81, 11749–11757. [Google Scholar] [CrossRef]
- Krzyżowska, M.; Cymerys, J.; Winnicka, A.; Niemiałtowski, M. Involvement of fas and fasL in ectromelia virus-induced apoptosis in mouse brain. Virus Res. 2006, 115, 141–149. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef]
- Pruitt, A.A. Central nervous system infections in cancer patients. Semin. Neurol. 2004, 24, 435–452. [Google Scholar] [CrossRef]
- Wang, Y.; Lobigs, M.; Lee, E.; Müllbacher, A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 2003, 77, 13323–13334. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M., Jr; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2’-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef]
- Stewart, B.S.; Demarest, V.L.; Wong, S.J.; Green, S.; Bernard, K.A. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 2011, 12, 6. [Google Scholar] [CrossRef]
- Appler, K.K.; Brown, A.N.; Stewart, B.S.; Behr, M.J.; Demarest, V.L.; Wong, S.J.; Bernard, K.A. Persistence of West Nile Virus in the central nervous system and periphery of mice. PLoS ONE 2010, 5, e10649. [Google Scholar]
- Slavin, H.B. Persistence of the virus of St. Louis encephalitis in the central nervous system of mice for over five months. J. Bacteriol. 1943, 46, 113–116. [Google Scholar]
- Pogodina, V.V.; Bochkova, N.G.; Levina, L.S. Persistence of tick-borne encephalitis virus in monkeys. VII. Some features of the immune response. Acta. Virol. 1984, 28, 407–415. [Google Scholar]
- Pogodina, V.V.; Frolova, M.P.; Malenko, G.V.; Fokina, G.I.; Levina, L.S.; Mamonenko, L.L.; Koreshkova, G.V.; Ralf, N.M. Persistence of tick-borne encephalitis virus in monkeys. I. Features of experimental infection. Acta. Virol. 1981, 25, 337–343. [Google Scholar]
- Zlotnik, I.; Carter, G.B.; Grant, D.P. The persistence of louping ill virus in immunosuppressed guinea-pigs. Br. J. Exp. Pathol. 1971, 52, 395–407. [Google Scholar]
- Pogodina, V.V.; Frolova, M.P.; Malenko, G.V.; Fokina, G.I.; Koreshkova, G.V.; Kiseleva, L.L.; Bochkova, N.G.; Ralph, N.M. Study on West Nile virus persistence in monkeys. Arch. Virol. 1983, 75, 71–86. [Google Scholar] [CrossRef]
- Xiao, S.Y.; Guzman, H.; Zhang, H.; Travassos da Rosa, A.P.; Tesh, R.B. West Nile virus infection in the golden hamster (Mesocricetus auratus): A model for West Nile encephalitis. Emerging Infect. Dis. 2001, 7, 714–721. [Google Scholar]
- Siddharthan, V.; Wang, H.; Motter, N.E.; Hall, J.O.; Skinner, R.D.; Skirpstunas, R.T.; Morrey, J.D. Persistent West Nile Virus associated with a neurological sequela in hamsters identified by motor unit number estimation. J. Virol. 2009, 83, 4251–4261. [Google Scholar] [CrossRef]
- Nolan, M.S.; Podoll, A.S.; Hause, A.M.; Akers, K.M.; Finkel, K.W.; Murray, K.O. Prevalence of chronic kidney disease and progression of disease over time among patients enrolled in the houston West Nile Virus cohort. PLoS ONE 2012, 7, e40374. [Google Scholar]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.C.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef]
- McCandless, E.E.; Zhang, B.; Diamond, M.S.; Klein, R.S. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. PNAS 2008, 105, 11270–11275. [Google Scholar] [CrossRef]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Müller, M.; Rana, S.; Deffrasnes, C.; Ashhurst, T.M.; Radford, J.; Hofer, M.; Thomas, S.; et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J. Neuroimmunol. 2012, 9, 246. [Google Scholar]
- Diamond, M.S. Progress on the development of therapeutics against West Nile virus. Antivir. Res. 2009, 83, 214–227. [Google Scholar]
- Dai, J.; Wang, P.; Bai, F.; Town, T.; Fikrig, E. ICAM-1 Participates in the entry of West Nile virus into the central nervous system. J. Virol. 2008, 82, 4164–4168. [Google Scholar] [CrossRef]
- Shen, J.; T-To, S.S.; Schrieber, L.; King, N.J. Early e-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J. Virol. 1997, 71, 9323–9332. [Google Scholar]
- Warke, R.V.; Martin, K.J.; Giaya, K.; Shaw, S.K.; Rothman, A.L.; Bosch, I. TRAIL Is a novel antiviral protein against Dengue virus. J. Virol. 2008, 82, 555–564. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cho, H.; Diamond, M.S. Immune Responses to West Nile Virus Infection in the Central Nervous System. Viruses 2012, 4, 3812-3830. https://doi.org/10.3390/v4123812
Cho H, Diamond MS. Immune Responses to West Nile Virus Infection in the Central Nervous System. Viruses. 2012; 4(12):3812-3830. https://doi.org/10.3390/v4123812
Chicago/Turabian StyleCho, Hyelim, and Michael S. Diamond. 2012. "Immune Responses to West Nile Virus Infection in the Central Nervous System" Viruses 4, no. 12: 3812-3830. https://doi.org/10.3390/v4123812
APA StyleCho, H., & Diamond, M. S. (2012). Immune Responses to West Nile Virus Infection in the Central Nervous System. Viruses, 4(12), 3812-3830. https://doi.org/10.3390/v4123812