Escape from Human Immunodeficiency Virus Type 1 (HIV-1) Entry Inhibitors

Abstract
:1. Introduction

2. Inhibitors of Envelope Glycoprotein-CD4 Receptor Interactions
2.1. Introduction

2.2. Protein Inhibitors of Env-Receptor Interactions
2.3. Small Molecule Inhibitors of Env-Receptor Interactions
2.4. Antibodies Targeting the CD4 Receptor
2.5. Summary
3. Inhibitors of Chemokine Receptor Interactions
3.1. Introduction
3.2. CXCR4 Inhibitors
3.3. CCR5 Inhibitors
3.3.1. Tropism Switch
3.3.2. Altered CCR5 Use
3.3.3. Resistance Mechanisms
3.4. Summary
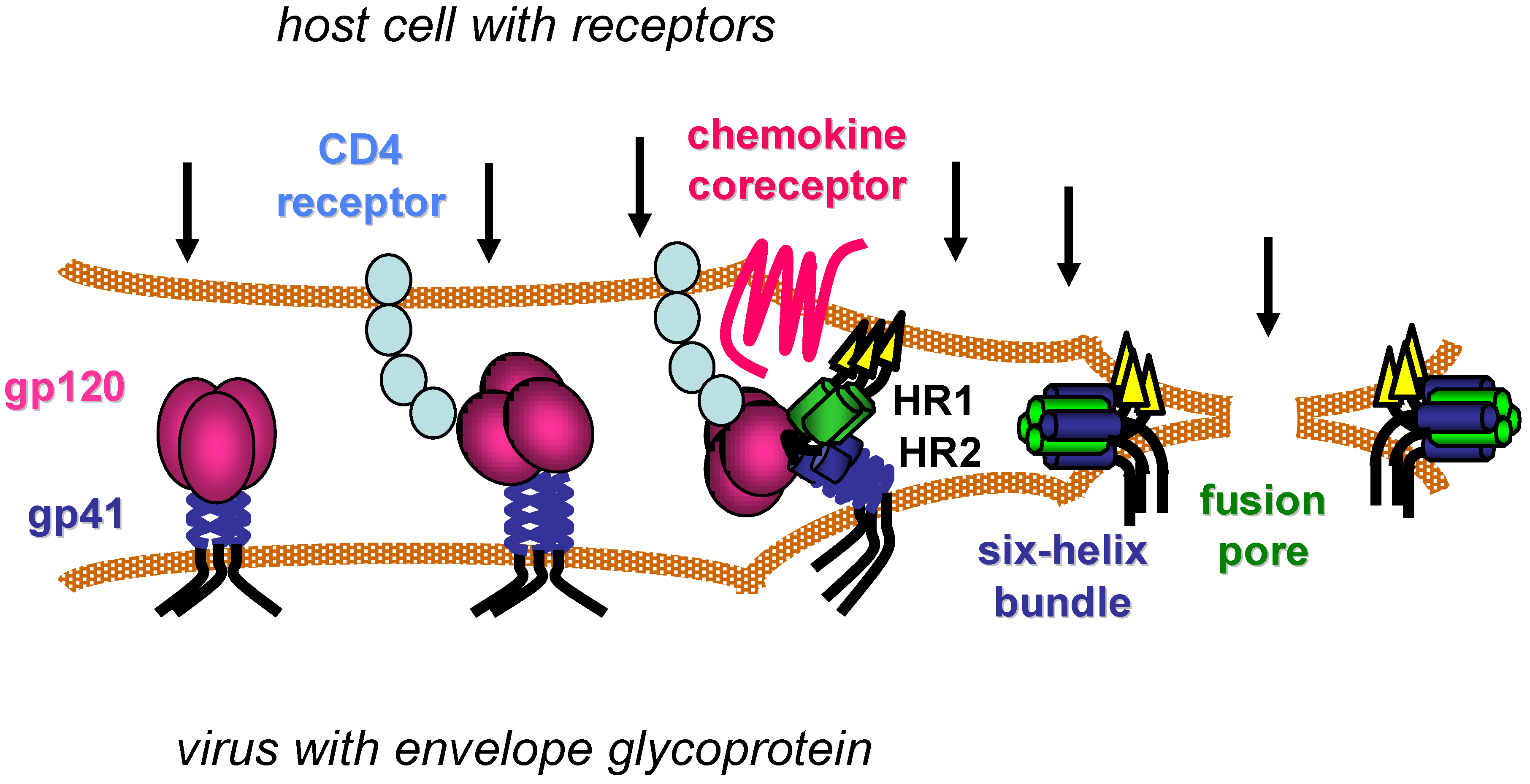
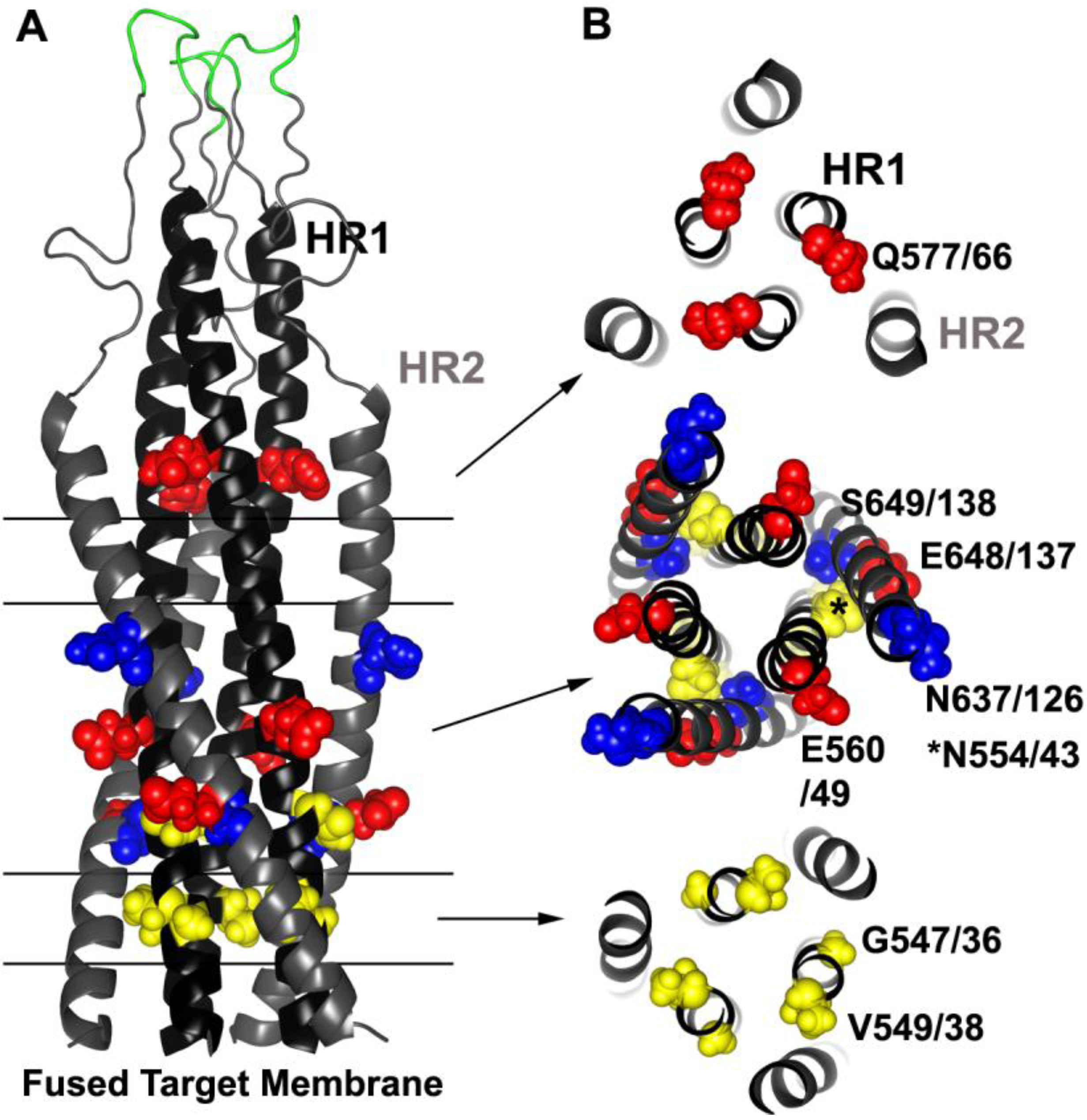
4. gp41 Fusion Inhibitors
4.1. Introduction
4.2. HR2 Fusion Inhibitors
4.2.1. T20


4.2.2. Next-Generation HR2 Peptides
4.3. HR1 Peptide Inhibitors

{kind=link}
{kind=link}
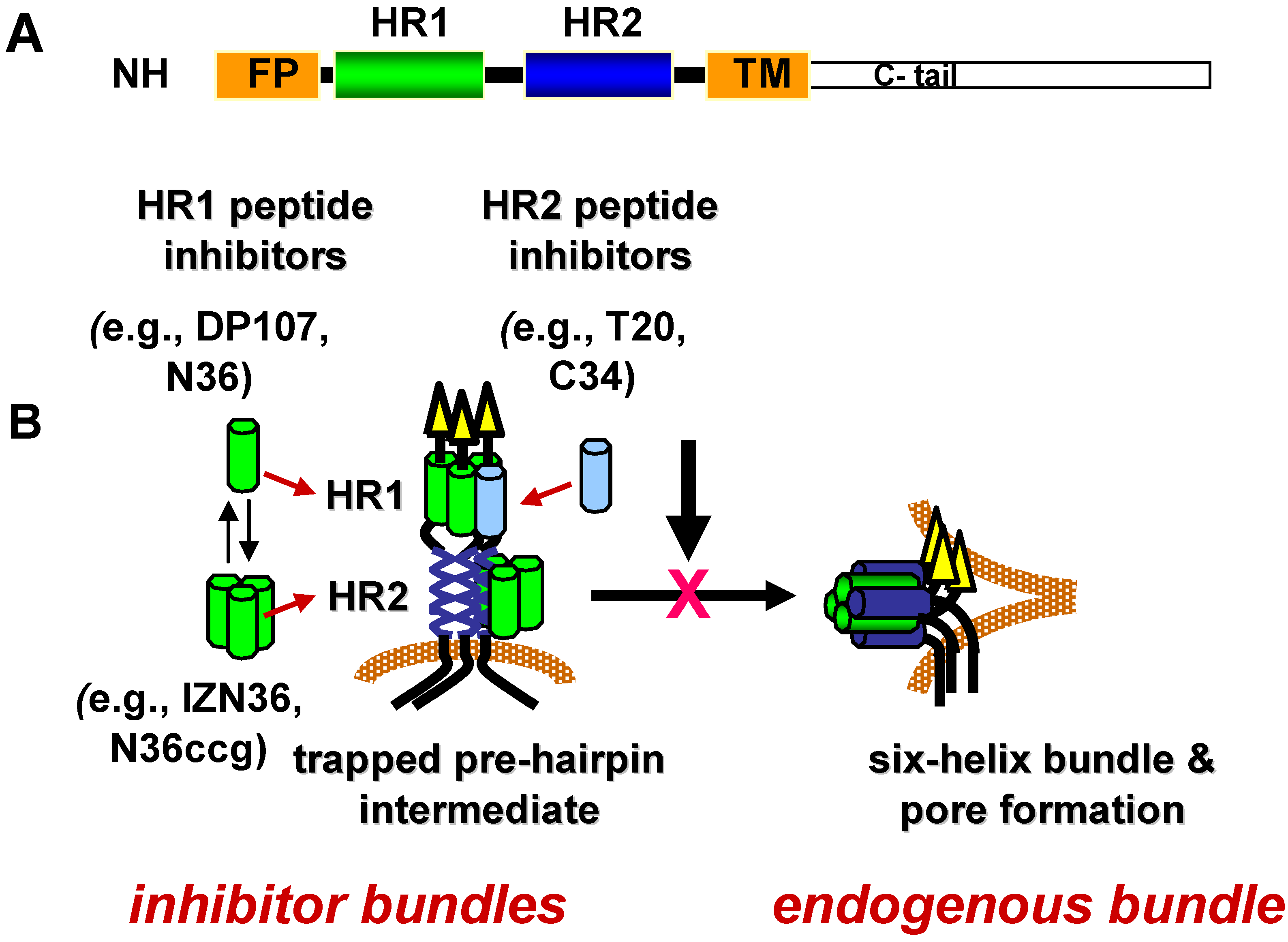{kind=link}
{kind=link}
 |
4.4. Summary
5. Other Entry Inhibitors
6. Final Perspective
Acknowledgements
References and Notes
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar]
- Antiretroviral drugs used in the treatment of HIV infection. Available online: http://www.fda.gov/forconsumers/byaudience/forpatientadvocates/hivandaidsactivities/ucm118915.htm (accessed on 6 November 2012).
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV outpatient study investigators. New Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef]
- Tsibris, A.M.; Hirsch, M.S. Antiretroviral therapy in the clinic. J. Virol. 2010, 84, 5458–5464. [Google Scholar] [CrossRef]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar]
- Wei, X.; Ghosh, S.K.; Taylor, M.E.; Johnson, V.A.; Emini, E.A.; Deutsch, P.; Lifson, J.D.; Bonhoeffer, S.; Nowak, M.A.; Hahn, B.H.; et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995, 373, 117–122. [Google Scholar]
- Tilton, J.C.; Doms, R.W. Entry inhibitors in the treatment of HIV-1 infection. Antivir. Res. 2010, 85, 91–100. [Google Scholar]
- Eggink, D.; Berkhout, B.; Sanders, R.W. Inhibition of HIV-1 by fusion inhibitors. Curr. Pharmaceut. Des. 2010, 16, 3716–3728. [Google Scholar] [CrossRef]
- Kuritzkes, D.R. HIV-1 entry inhibitors: An overview. Curr. Opin. HIV AIDS 2009, 4, 82–87. [Google Scholar] [CrossRef]
- Cai, L.; Jiang, S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem 2010, 5, 1813–1824. [Google Scholar] [CrossRef]
- Ugolini, S.; Mondor, I.; Sattentau, Q.J. HIV-1 attachment: Another look. Trends Microbiol. 1999, 7, 144–149. [Google Scholar] [CrossRef]
- Klatzmann, D.; Champagne, E.; Chamaret, S.; Gruest, J.; Guetard, D.; Hercend, T.; Gluckman, J.C.; Montagnier, L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 1984, 312, 767–768. [Google Scholar]
- Dalgleish, A.G.; Beverley, P.C.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312, 763–767. [Google Scholar]
- McDougal, J.S.; Kennedy, M.S.; Sligh, J.M.; Cort, S.P.; Mawle, A.; Nicholson, J.K. Binding of HTLV-III/LAV to T4+ T cells by a complex of the 110K viral protein and the T4 molecule. Science 1986, 231, 382–385. [Google Scholar]
- Maddon, P.J.; Dalgleish, A.G.; McDougal, J.S.; Clapham, P.R.; Weiss, R.A.; Axel, R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986, 47, 333–348. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Zaitseva, M.; Peden, K.; Golding, H. HIV coreceptors: Role of structure, posttranslational modifications, and internalization in viral-cell fusion and as targets for entry inhibitors. Biochim. Biophys. Acta 2003, 1614, 51–61. [Google Scholar]
- Melikyan, G.B. Membrane fusion mediated by human immunodeficiency virus envelope glycoprotein. Curr. Top. Membr. 2011, 68, 81–106. [Google Scholar] [CrossRef]
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Moore, J.P.; McKeating, J.A.; Weiss, R.A.; Sattentau, Q.J. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 1990, 250, 1139–1142. [Google Scholar]
- Sattentau, Q.J.; Moore, J.P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415. [Google Scholar] [CrossRef]
- Wu, L.; Gerard, N.P.; Wyatt, R.; Choe, H.; Parolin, C.; Ruffing, N.; Borsetti, A.; Cardoso, A.A.; Desjardin, E.; Newman, W.; et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384, 179–183. [Google Scholar]
- Trkola, A.; Dragic, T.; Arthos, J.; Binley, J.M.; Olson, W.C.; Allaway, G.P.; Cheng-Mayer, C.; Robinson, J.; Maddon, P.J.; Moore, J.P. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 1996, 384, 184–187. [Google Scholar]
- Thali, M.; Moore, J.P.; Furman, C.; Charles, M.; Ho, D.D.; Robinson, J.; Sodroski, J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 1993, 67, 3978–3988. [Google Scholar]
- Chen, B.; Vogan, E.M.; Gong, H.; Skehel, J.J.; Wiley, D.C.; Harrison, S.C. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 2005, 433, 834–841. [Google Scholar]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar]
- Myszka, D.G.; Sweet, R.W.; Hensley, P.; Brigham-Burke, M.; Kwong, P.D.; Hendrickson, W.A.; Wyatt, R.; Sodroski, J.; Doyle, M.L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9026–9031. [Google Scholar]
- Kwon, Y.D.; Finzi, A.; Wu, X.; Dogo-Isonagie, C.; Lee, L.K.; Moore, L.R.; Schmidt, S.D.; Stuckey, J.; Yang, Y.; Zhou, T.; et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5663–5668. [Google Scholar]
- Yuan, W.; Bazick, J.; Sodroski, J. Characterization of the multiple conformational States of free monomeric and trimeric human immunodeficiency virus envelope glycoproteins after fixation by cross-linker. J. Virol. 2006, 80, 6725–6737. [Google Scholar] [CrossRef]
- Kong, L.; Huang, C.C.; Coales, S.J.; Molnar, K.S.; Skinner, J.; Hamuro, Y.; Kwong, P.D. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J. Virol. 2010, 84, 10311–10321. [Google Scholar]
- Kwong, P.D.; Doyle, M.L.; Casper, D.J.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [Google Scholar]
- Xiang, S.H.; Kwong, P.D.; Gupta, R.; Rizzuto, C.D.; Casper, D.J.; Wyatt, R.; Wang, L.; Hendrickson, W.A.; Doyle, M.L.; Sodroski, J. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 2002, 76, 9888–9899. [Google Scholar]
- Pancera, M.; Majeed, S.; Ban, Y.E.; Chen, L.; Huang, C.C.; Kong, L.; Kwon, Y.D.; Stuckey, J.; Zhou, T.; Robinson, J.E.; et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1166–1171. [Google Scholar]
- Huang, C.C.; Tang, M.; Zhang, M.Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; et al. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar]
- Huang, C.C.; Lam, S.N.; Acharya, P.; Tang, M.; Xiang, S.H.; Hussan, S.S.; Stanfield, R.L.; Robinson, J.; Sodroski, J.; Wilson, I.A.; et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 2007, 317, 1930–1934. [Google Scholar] [CrossRef]
- Hussey, R.E.; Richardson, N.E.; Kowalski, M.; Brown, N.R.; Chang, H.C.; Siliciano, R.F.; Dorfman, T.; Walker, B.; Sodroski, J.; Reinherz, E.L. A soluble CD4 protein selectively inhibits HIV replication and syncytium formation. Nature 1988, 331, 78–81. [Google Scholar]
- Deen, K.C.; McDougal, J.S.; Inacker, R.; Folena-Wasserman, G.; Arthos, J.; Rosenberg, J.; Maddon, P.J.; Axel, R.; Sweet, R.W. A soluble form of CD4 (T4) protein inhibits AIDS virus infection. Nature 1988, 331, 82–84. [Google Scholar]
- Fisher, R.A.; Bertonis, J.M.; Meier, W.; Johnson, V.A.; Costopoulos, D.S.; Liu, T.; Tizard, R.; Walker, B.D.; Hirsch, M.S.; Schooley, R.T.; et al. HIV infection is blocked in vitro by recombinant soluble CD4. Nature 1988, 331, 76–78. [Google Scholar]
- Clapham, P.R.; Weber, J.N.; Whitby, D.; McIntosh, K.; Dalgleish, A.G.; Maddon, P.J.; Deen, K.C.; Sweet, R.W.; Weiss, R.A. Soluble CD4 blocks the infectivity of diverse strains of HIV and SIV for T cells and monocytes but not for brain and muscle cells. Nature 1989, 337, 368–370. [Google Scholar]
- Allan, J.S.; Strauss, J.; Buck, D.W. Enhancement of SIV infection with soluble receptor molecules. Science 1990, 247, 1084–1088. [Google Scholar]
- Clapham, P.R.; McKnight, A.; Weiss, R.A. Human immunodeficiency virus type 2 infection and fusion of CD4-negative human cell lines: Induction and enhancement by soluble CD4. J. Virol. 1992, 66, 3531–3537. [Google Scholar]
- Allan, J.S.; Whitehead, E.M.; Strout, K.; Short, M.; Kanda, P.; Hart, T.K.; Bugelski, P.J. Strong association of simian immunodeficiency virus (SIVagm) envelope glycoprotein heterodimers: Possible role in receptor-mediated activation. AIDS Res. Hum. Retrovir. 1992, 8, 2011–2020. [Google Scholar] [CrossRef]
- Sullivan, N.; Sun, Y.; Li, J.; Hofmann, W.; Sodroski, J. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line-passaged human immunodeficiency virus type 1 isolates. J. Virol. 1995, 69, 4413–4422. [Google Scholar]
- Schutten, M.; Andeweg, A.C.; Bosch, M.L.; Osterhaus, A.D. Enhancement of infectivity of a non-syncytium inducing HIV-1 by sCD4 and by human antibodies that neutralize syncytium inducing HIV-1. Scand. J. Immunol. 1995, 41, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Daar, E.S.; Li, X.L.; Moudgil, T.; Ho, D.D. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 6574–6578. [Google Scholar] [CrossRef]
- Brighty, D.W.; Rosenberg, M.; Chen, I.S.; Ivey-Hoyle, M. Envelope proteins from clinical isolates of human immunodeficiency virus type 1 that are refractory to neutralization by soluble CD4 possess high affinity for the CD4 receptor. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 7802–7805. [Google Scholar]
- Moore, J.P.; McKeating, J.A.; Huang, Y.X.; Ashkenazi, A.; Ho, D.D. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J. Virol. 1992, 66, 235–243. [Google Scholar]
- Turner, S.; Tizard, R.; DeMarinis, J.; Pepinsky, R.B.; Zullo, J.; Schooley, R.; Fisher, R. Resistance of primary isolates of human immunodeficiency virus type 1 to neutralization by soluble CD4 is not due to lower affinity with the viral envelope glycoprotein gp120. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 1335–1339. [Google Scholar]
- Orloff, S.L.; Kennedy, M.S.; Belperron, A.A.; Maddon, P.J.; McDougal, J.S. Two mechanisms of soluble CD4 (sCD4)-mediated inhibition of human immunodeficiency virus type 1 (HIV-1) infectivity and their relation to primary HIV-1 isolates with reduced sensitivity to sCD4. J. Virol. 1993, 67, 1461–1471. [Google Scholar]
- Groenink, M.; Moore, J.P.; Broersen, S.; Schuitemaker, H. Equal levels of gp120 retention and neutralization resistance of phenotypically distinct primary human immunodeficiency virus type 1 variants upon soluble CD4 treatment. J. Virol. 1995, 69, 523–527. [Google Scholar]
- Moore, J.P.; McKeating, J.A.; Norton, W.A.; Sattentau, Q.J. Direct measurement of soluble CD4 binding to human immunodeficiency virus type 1 virions: gp120 dissociation and its implications for virus-cell binding and fusion reactions and their neutralization by soluble CD4. J. Virol. 1991, 65, 1133–1140. [Google Scholar]
- Hart, T.K.; Kirsh, R.; Ellens, H.; Sweet, R.W.; Lambert, D.M.; Petteway, S.R., Jr.; Leary, J.; Bugelski, P.J. Binding of soluble CD4 proteins to human immunodeficiency virus type 1 and infected cells induces release of envelope glycoprotein gp120. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 2189–2193. [Google Scholar]
- Vita, C.; Drakopoulou, E.; Vizzavona, J.; Rochette, S.; Martin, L.; Menez, A.; Roumestand, C.; Yang, Y.S.; Ylisastigui, L.; Benjouad, A.; et al. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 13091–13096. [Google Scholar]
- Martin, L.; Stricher, F.; Misse, D.; Sironi, F.; Pugniere, M.; Barthe, P.; Prado-Gotor, R.; Freulon, I.; Magne, X.; Roumestand, C.; et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 2003, 21, 71–76. [Google Scholar] [CrossRef]
- Stricher, F.; Huang, C.C.; Descours, A.; Duquesnoy, S.; Combes, O.; Decker, J.M.; Kwon, Y.D.; Lusso, P.; Shaw, G.M.; Vita, C.; et al. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J. Mol. Biol. 2008, 382, 510–524. [Google Scholar] [CrossRef]
- Grupping, K.; Selhorst, P.; Michiels, J.; Vereecken, K.; Heyndrickx, L.; Kessler, P.; Vanham, G.; Martin, L.; Arien, K.K. MiniCD4 protein resistance mutations affect binding to the HIV-1 gp120 CD4 binding site and decrease entry efficiency. Retrovirology 2012, 9, 36. [Google Scholar] [CrossRef]
- Yoshimura, K.; Harada, S.; Shibata, J.; Hatada, M.; Yamada, Y.; Ochiai, C.; Tamamura, H.; Matsushita, S. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J. Virol. 2010, 84, 7558–7568. [Google Scholar]
- McKeating, J.; Balfe, P.; Clapham, P.; Weiss, R.A. Recombinant CD4-selected human immunodeficiency virus type 1 variants with reduced gp120 affinity for CD4 and increased cell fusion capacity. J. Virol. 1991, 65, 4777–4785. [Google Scholar]
- McKeating, J.A.; Bennett, J.; Zolla-Pazner, S.; Schutten, M.; Ashelford, S.; Brown, A.L.; Balfe, P. Resistance of a human serum-selected human immunodeficiency virus type 1 escape mutant to neutralization by CD4 binding site monoclonal antibodies is conferred by a single amino acid change in gp120. J. Virol. 1993, 67, 5216–5225. [Google Scholar]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N'-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef]
- Schon, A.; Madani, N.; Klein, J.C.; Hubicki, A.; Ng, D.; Yang, X.; Smith, A.B., 3rd; Sodroski, J.; Freire, E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 2006, 45, 10973–10980. [Google Scholar]
- Madani, N.; Schon, A.; Princiotto, A.M.; Lalonde, J.M.; Courter, J.R.; Soeta, T.; Ng, D.; Wang, L.; Brower, E.T.; Xiang, S.H.; et al. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 2008, 16, 1689–1701. [Google Scholar] [CrossRef]
- Yoshimura, K.; Harada, S.; Shibata, J.; Hatada, M.; Yamada, Y.; Ochiai, C.; Tamamura, H.; Matsushita, S. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J. Virol. 84 7558–7568.
- Yamada, Y.; Ochiai, C.; Yoshimura, K.; Tanaka, T.; Ohashi, N.; Narumi, T.; Nomura, W.; Harada, S.; Matsushita, S.; Tamamura, H. CD4 mimics targeting the mechanism of HIV entry. Bioorg. Med. Chem. Lett. 2010, 20, 354–358. [Google Scholar] [CrossRef]
- Haim, H.; Si, Z.; Madani, N.; Wang, L.; Courter, J.R.; Princiotto, A.; Kassa, A.; DeGrace, M.; McGee-Estrada, K.; Mefford, M.; et al. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 2009, 5, e1000360. [Google Scholar] [CrossRef]
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.; Rose, R.; Yamanaka, G.; et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11013–11018. [Google Scholar]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef]
- Madani, N.; Perdigoto, A.L.; Srinivasan, K.; Cox, J.M.; Chruma, J.J.; LaLonde, J.; Head, M.; Smith, A.B., 3rd; Sodroski, J.G. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 2004, 78, 3742–3752. [Google Scholar] [CrossRef]
- Zhou, N.; Nowicka-Sans, B.; Zhang, S.; Fan, L.; Fang, J.; Fang, H.; Gong, Y.F.; Eggers, B.; Langley, D.R.; Wang, T.; et al. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob. Agents Chemother. 2011, 55, 729–737. [Google Scholar] [CrossRef]
- Guo, Q.; Ho, H.T.; Dicker, I.; Fan, L.; Zhou, N.; Friborg, J.; Wang, T.; McAuliffe, B.V.; Wang, H.G.; Rose, R.E.; et al. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J. Virol. 2003, 77, 10528–10536. [Google Scholar]
- Si, Z.; Madani, N.; Cox, J.M.; Chruma, J.J.; Klein, J.C.; Schon, A.; Phan, N.; Wang, L.; Biorn, A.C.; Cocklin, S.; et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5036–5041. [Google Scholar]
- Ho, H.T.; Fan, L.; Nowicka-Sans, B.; McAuliffe, B.; Li, C.B.; Yamanaka, G.; Zhou, N.; Fang, H.; Dicker, I.; Dalterio, R.; et al. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J. Virol. 2006, 80, 4017–4025. [Google Scholar] [CrossRef]
- Zhou, N.; Fan, L.; Ho, H.T.; Nowicka-Sans, B.; Sun, Y.; Zhu, Y.; Hu, Y.; McAuliffe, B.; Rose, B.; Fang, H.; et al. Increased sensitivity of HIV variants selected by attachment inhibitors to broadly neutralizing antibodies. Virology 2010, 402, 256–261. [Google Scholar] [CrossRef]
- Vermeire, K.; Zhang, Y.; Princen, K.; Hatse, S.; Samala, M.F.; Dey, K.; Choi, H.J.; Ahn, Y.; Sodoma, A.; Snoeck, R.; et al. CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology 2002, 302, 342–353. [Google Scholar] [CrossRef]
- Vermeire, K.; Schols, D. Cyclotriazadisulfonamides: Promising new CD4-targeted anti-HIV drugs. J. Antimicrob. Chemother. 2005, 56, 270–272. [Google Scholar] [CrossRef]
- Vermeire, K.; Bell, T.W.; Choi, H.J.; Jin, Q.; Samala, M.F.; Sodoma, A.; de Clercq, E.; Schols, D. The anti-HIV potency of cyclotriazadisulfonamide analogs is directly correlated with their ability to down-modulate the CD4 receptor. Mol. Pharmacol. 2003, 63, 203–210. [Google Scholar] [CrossRef]
- Kabat, D.; Kozak, S.L.; Wehrly, K.; Chesebro, B. Differences in CD4 dependence for infectivity of laboratory-adapted and primary patient isolates of human immunodeficiency virus type 1. J. Virol. 1994, 68, 2570–2577. [Google Scholar]
- Vermeire, K.; Van Laethem, K.; Janssens, W.; Bell, T.W.; Schols, D. Human immunodeficiency virus type 1 escape from cyclotriazadisulfonamide-induced CD4-targeted entry inhibition is associated with increased neutralizing antibody susceptibility. J. Virol. 2009, 83, 9577–9583. [Google Scholar] [CrossRef]
- Vermeire, K.; Brouwers, J.; Van Herrewege, Y.; Le Grand, R.; Vanham, G.; Augustijns, P.; Bell, T.W.; Schols, D. CADA, a potential anti-HIV microbicide that specifically targets the cellular CD4 receptor. Curr. HIV Res. 2008, 6, 246–256. [Google Scholar] [CrossRef]
- Reimann, K.A.; Lin, W.; Bixler, S.; Browning, B.; Ehrenfels, B.N.; Lucci, J.; Miatkowski, K.; Olson, D.; Parish, T.H.; Rosa, M.D.; et al. A humanized form of a CD4-specific monoclonal antibody exhibits decreased antigenicity and prolonged plasma half-life in rhesus monkeys while retaining its unique biological and antiviral properties. AIDS Res. Hum. Retrovir. 1997, 13, 933–943. [Google Scholar] [CrossRef]
- Burkly, L.C.; Olson, D.; Shapiro, R.; Winkler, G.; Rosa, J.J.; Thomas, D.W.; Williams, C.; Chisholm, P. Inhibition of HIV infection by a novel CD4 domain 2-specific monoclonal antibody. Dissecting the basis for its inhibitory effect on HIV-induced cell fusion. J. Immunol. 1992, 149, 1779–1787. [Google Scholar]
- Moore, J.P.; Sattentau, Q.J.; Klasse, P.J.; Burkly, L.C. A monoclonal antibody to CD4 domain 2 blocks soluble CD4-induced conformational changes in the envelope glycoproteins of human immunodeficiency virus type 1 (HIV-1) and HIV-1 infection of CD4+ cells. J. Virol. 1992, 66, 4784–4793. [Google Scholar]
- Reimann, K.A.; Burkly, L.C.; Burrus, B.; Waite, B.C.; Lord, C.I.; Letvin, N.L. In vivo administration to rhesus monkeys of a CD4-specific monoclonal antibody capable of blocking AIDS virus replication. AIDS Res. Hum. Retrovir. 1993, 9, 199–207. [Google Scholar] [CrossRef]
- Boon, L.; Holland, B.; Gordon, W.; Liu, P.; Shiau, F.; Shanahan, W.; Reimann, K.A.; Fung, M. Development of anti-CD4 MAb hu5A8 for treatment of HIV-1 infection: Preclinical assessment in non-human primates. Toxicology 2002, 172, 191–203. [Google Scholar] [CrossRef]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.; Kao, C.Y.; Ho, D.D.; Chen, B. Crystal structure of HIV-1 primary receptor CD4 in complex with a potent antiviral antibody. Structure 2010, 18, 1632–1641. [Google Scholar] [CrossRef]
- Reimann, K.A.; Khunkhun, R.; Lin, W.; Gordon, W.; Fung, M. A humanized, nondepleting anti-CD4 antibody that blocks virus entry inhibits virus replication in rhesus monkeys chronically infected with simian immunodeficiency virus. AIDS Res. Hum. Retrovir. 2002, 18, 747–755. [Google Scholar] [CrossRef]
- Jacobson, J.M.; Kuritzkes, D.R.; Godofsky, E.; DeJesus, E.; Larson, J.A.; Weinheimer, S.P.; Lewis, S.T. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob. Agents Chemother. 2009, 53, 450–457. [Google Scholar] [CrossRef]
- Kuritzkes, D.R.; Jacobson, J.; Powderly, W.G.; Godofsky, E.; DeJesus, E.; Haas, F.; Reimann, K.A.; Larson, J.L.; Yarbough, P.O.; Curt, V.; et al. Antiretroviral activity of the anti-CD4 monoclonal antibody TNX-355 in patients infected with HIV type 1. J. Infect. Dis. 2004, 189, 286–291. [Google Scholar] [CrossRef]
- Toma, J.; Weinheimer, S.P.; Stawiski, E.; Whitcomb, J.M.; Lewis, S.T.; Petropoulos, C.J.; Huang, W. Loss of asparagine-linked glycosylation sites in variable region 5 of human immunodeficiency virus type 1 envelope is associated with resistance to CD4 antibody ibalizumab. J. Virol. 2011, 85, 3872–3880. [Google Scholar] [CrossRef]
- Tran, E.E.; Borgnia, M.J.; Kuybeda, O.; Schauder, D.M.; Bartesaghi, A.; Frank, G.A.; Sapiro, G.; Milne, J.L.; Subramaniam, S. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 2012, 8, e1002797. [Google Scholar] [CrossRef]
- Gallo, S.A.; Finnegan, C.M.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 2003, 1614, 36–50. [Google Scholar]
- Berson, J.F.; Long, D.; Doranz, B.J.; Rucker, J.; Jirik, F.R.; Doms, R.W. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strains. J. Virol. 1996, 70, 6288–6295. [Google Scholar]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar]
- Bleul, C.C.; Farzan, M.; Choe, H.; Parolin, C.; Clark-Lewis, I.; Sodroski, J.; Springer, T.A. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996, 382, 829–833. [Google Scholar]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996, 382, 833–835. [Google Scholar]
- Rizzuto, C.D.; Wyatt, R.; Hernandez-Ramos, N.; Sun, Y.; Kwong, P.D.; Hendrickson, W.A.; Sodroski, J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 1998, 280, 1949–1953. [Google Scholar] [CrossRef]
- Rizzuto, C.; Sodroski, J. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retrovir. 2000, 16, 741–749. [Google Scholar] [CrossRef]
- Doranz, B.J.; Lu, Z.H.; Rucker, J.; Zhang, T.Y.; Sharron, M.; Cen, Y.H.; Wang, Z.X.; Guo, H.H.; Du, J.G.; Accavitti, M.A.; et al. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J. Virol. 1997, 71, 6305–6314. [Google Scholar]
- Farzan, M.; Choe, H.; Vaca, L.; Martin, K.; Sun, Y.; Desjardins, E.; Ruffing, N.; Wu, L.; Wyatt, R.; Gerard, N.; et al. A tyrosine-rich region in the N terminus of CCR5 is important for human immunodeficiency virus type 1 entry and mediates an association between gp120 and CCR5. J. Virol. 1998, 72, 1160–1164. [Google Scholar]
- Kajumo, F.; Thompson, D.A.; Guo, Y.; Dragic, T. Entry of R5X4 and X4 human immunodeficiency virus type 1 strains is mediated by negatively charged and tyrosine residues in the amino-terminal domain and the second extracellular loop of CXCR4. Virology 2000, 271, 240–247. [Google Scholar] [CrossRef]
- Cormier, E.G.; Tran, D.N.; Yukhayeva, L.; Olson, W.C.; Dragic, T. Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J. Virol. 2001, 75, 5541–5549. [Google Scholar] [CrossRef]
- Cormier, E.G.; Dragic, T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 2002, 76, 8953–8957. [Google Scholar] [CrossRef]
- Nolan, K.M.; Jordan, A.P.; Hoxie, J.A. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J. Virol. 2008, 82, 664–673. [Google Scholar] [CrossRef]
- Lee, B.; Sharron, M.; Blanpain, C.; Doranz, B.J.; Vakili, J.; Setoh, P.; Berg, E.; Liu, G.; Guy, H.R.; Durell, S.R.; et al. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 1999, 274, 9617–9626. [Google Scholar]
- Farzan, M.; Mirzabekov, T.; Kolchinsky, P.; Wyatt, R.; Cayabyab, M.; Gerard, N.P.; Gerard, C.; Sodroski, J.; Choe, H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 1999, 96, 667–676. [Google Scholar] [CrossRef]
- Arenzana-Seisdedos, F.; Virelizier, J.L.; Rousset, D.; Clark-Lewis, I.; Loetscher, P.; Moser, B.; Baggiolini, M. HIV blocked by chemokine antagonist. Nature 1996, 383, 400. [Google Scholar] [CrossRef]
- Amara, A.; Gall, S.L.; Schwartz, O.; Salamero, J.; Montes, M.; Loetscher, P.; Baggiolini, M.; Virelizier, J.L.; Arenzana-Seisdedos, F. HIV coreceptor downregulation as antiviral principle: SDF-1alpha-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J. Exp. Med. 1997, 186, 139–146. [Google Scholar] [CrossRef]
- Signoret, N.; Oldridge, J.; Pelchen-Matthews, A.; Klasse, P.J.; Tran, T.; Brass, L.F.; Rosenkilde, M.M.; Schwartz, T.W.; Holmes, W.; Dallas, W.; et al. Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J. Cell Biol. 1997, 139, 651–664. [Google Scholar] [CrossRef]
- Simmons, G.; Clapham, P.R.; Picard, L.; Offord, R.E.; Rosenkilde, M.M.; Schwartz, T.W.; Buser, R.; Wells, T.N.; Proudfoot, A.E. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 1997, 276, 276–279. [Google Scholar] [CrossRef]
- Mack, M.; Luckow, B.; Nelson, P.J.; Cihak, J.; Simmons, G.; Clapham, P.R.; Signoret, N.; Marsh, M.; Stangassinger, M.; Borlat, F.; et al. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: A novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 1998, 187, 1215–1224. [Google Scholar] [CrossRef]
- Pastore, C.; Picchio, G.R.; Galimi, F.; Fish, R.; Hartley, O.; Offord, R.E.; Mosier, D.E. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 2003, 47, 509–517. [Google Scholar] [CrossRef]
- Baba, M.; Nishimura, O.; Kanzaki, N.; Okamoto, M.; Sawada, H.; Iizawa, Y.; Shiraishi, M.; Aramaki, Y.; Okonogi, K.; Ogawa, Y.; et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5698–5703. [Google Scholar]
- Dragic, T.; Trkola, A.; Thompson, D.A.; Cormier, E.G.; Kajumo, F.A.; Maxwell, E.; Lin, S.W.; Ying, W.; Smith, S.O.; Sakmar, T.P.; et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5639–5644. [Google Scholar]
- Strizki, J.M.; Xu, S.; Wagner, N.E.; Wojcik, L.; Liu, J.; Hou, Y.; Endres, M.; Palani, A.; Shapiro, S.; Clader, J.W.; et al. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12718–12723. [Google Scholar]
- Kazmierski, W.; Bifulco, N.; Yang, H.; Boone, L.; DeAnda, F.; Watson, C.; Kenakin, T. Recent progress in discovery of small-molecule CCR5 chemokine receptor ligands as HIV-1 inhibitors. Bioorg. Med. Chem. 2003, 11, 2663–2676. [Google Scholar] [CrossRef]
- Tsamis, F.; Gavrilov, S.; Kajumo, F.; Seibert, C.; Kuhmann, S.; Ketas, T.; Trkola, A.; Palani, A.; Clader, J.W.; Tagat, J.R.; et al. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 2003, 77, 5201–5208. [Google Scholar]
- Billick, E.; Seibert, C.; Pugach, P.; Ketas, T.; Trkola, A.; Endres, M.J.; Murgolo, N.J.; Coates, E.; Reyes, G.R.; Baroudy, B.M.; et al. The differential sensitivity of human and rhesus macaque CCR5 to small-molecule inhibitors of human immunodeficiency virus type 1 entry is explained by a single amino acid difference and suggests a mechanism of action for these inhibitors. J. Virol. 2004, 78, 4134–4144. [Google Scholar]
- Maeda, K.; Nakata, H.; Koh, Y.; Miyakawa, T.; Ogata, H.; Takaoka, Y.; Shibayama, S.; Sagawa, K.; Fukushima, D.; Moravek, J.; et al. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J. Virol. 2004, 78, 8654–8662. [Google Scholar] [CrossRef]
- Baba, M.; Takashima, K.; Miyake, H.; Kanzaki, N.; Teshima, K.; Wang, X.; Shiraishi, M.; Iizawa, Y. TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans. Antimicrob. Agents Chemother. 2005, 49, 4584–4591. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar]
- Takashima, K.; Miyake, H.; Kanzaki, N.; Tagawa, Y.; Wang, X.; Sugihara, Y.; Iizawa, Y.; Baba, M. Highly potent inhibition of human immunodeficiency virus type 1 replication by TAK-220, an orally bioavailable small-molecule CCR5 antagonist. Antimicrob. Agents Chemother. 2005, 49, 3474–3482. [Google Scholar] [CrossRef]
- Watson, C.; Jenkinson, S.; Kazmierski, W.; Kenakin, T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol. Pharmacol. 2005, 67, 1268–1282. [Google Scholar] [CrossRef]
- Seibert, C.; Ying, W.; Gavrilov, S.; Tsamis, F.; Kuhmann, S.E.; Palani, A.; Tagat, J.R.; Clader, J.W.; McCombie, S.W.; Baroudy, B.M.; et al. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology 2006, 349, 41–54. [Google Scholar] [CrossRef]
- Maeda, K.; Das, D.; Yin, P.D.; Tsuchiya, K.; Ogata-Aoki, H.; Nakata, H.; Norman, R.B.; Hackney, L.A.; Takaoka, Y.; Mitsuya, H. Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV-1 fusion: Insights into the mechanism of allosteric inhibition. J. Mol. Biol. 2008, 381, 956–974. [Google Scholar] [CrossRef]
- Muniz-Medina, V.M.; Jones, S.; Maglich, J.M.; Galardi, C.; Hollingsworth, R.E.; Kazmierski, W.M.; Ferris, R.G.; Edelstein, M.P.; Chiswell, K.E.; Kenakin, T.P. The relative activity of "function sparing" HIV-1 entry inhibitors on viral entry and CCR5 internalization: Is allosteric functional selectivity a valuable therapeutic property? Mol. Pharmacol. 2009, 75, 490–501. [Google Scholar] [CrossRef]
- Tilton, J.C.; Wilen, C.B.; Didigu, C.A.; Sinha, R.; Harrison, J.E.; Agrawal-Gamse, C.; Henning, E.A.; Bushman, F.D.; Martin, J.N.; Deeks, S.G.; et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 2010, 84, 10863–10876. [Google Scholar]
- Garcia-Perez, J.; Rueda, P.; Staropoli, I.; Kellenberger, E.; Alcami, J.; Arenzana-Seisdedos, F.; Lagane, B. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem. 2011, 286, 4978–4990. [Google Scholar]
- Maeda, K.; Das, D.; Ogata-Aoki, H.; Nakata, H.; Miyakawa, T.; Tojo, Y.; Norman, R.; Takaoka, Y.; Ding, J.; Arnold, G.F.; et al. Structural and molecular interactions of CCR5 inhibitors with CCR5. J. Biol. Chem. 2006, 281, 12688–12698. [Google Scholar]
- Kondru, R.; Zhang, J.; Ji, C.; Mirzadegan, T.; Rotstein, D.; Sankuratri, S.; Dioszegi, M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 2008, 73, 789–800. [Google Scholar]
- Gilliam, B.L.; Riedel, D.J.; Redfield, R.R. Clinical use of CCR5 inhibitors in HIV and beyond. J. Trans.l Med. 2011, 9, S9. [Google Scholar]
- Kanbara, K.; Sato, S.; Tanuma, J.; Tamamura, H.; Gotoh, K.; Yoshimori, M.; Kanamoto, T.; Kitano, M.; Fujii, N.; Nakashima, H. Biological and genetic characterization of a human immunodeficiency virus strain resistant to CXCR4 antagonist T134. AIDS Res. Hum. Retrovir. 2001, 17, 615–622. [Google Scholar] [CrossRef]
- Schols, D.; Este, J.A.; Cabrera, C.; de Clercq, E. T-cell-line-tropic human immunodeficiency virus type 1 that is made resistant to stromal cell-derived factor 1alpha contains mutations in the envelope gp120 but does not show a switch in coreceptor use. J. Virol. 1998, 72, 4032–4037. [Google Scholar]
- De Vreese, K.; Kofler-Mongold, V.; Leutgeb, C.; Weber, V.; Vermeire, K.; Schacht, S.; Anne, J.; de Clercq, E.; Datema, R.; Werner, G. The molecular target of bicyclams, potent inhibitors of human immunodeficiency virus replication. J. Virol. 1996, 70, 689–696. [Google Scholar]
- Este, J.A.; de Vreese, K.; Witvrouw, M.; Schmit, J.C.; Vandamme, A.M.; Anne, J.; Desmyter, J.; Henson, G.W.; Bridger, G.; de Clercq, E. Antiviral activity of the bicyclam derivative JM3100 against drug-resistant strains of human immunodeficiency virus type 1. Antivir. Res. 1996, 29, 297–307. [Google Scholar]
- Este, J.A.; Schols, D.; de Vreese, K.; van Laethem, K.; Vandamme, A.M.; Desmyter, J.; de Clercq, E. Development of resistance of human immunodeficiency virus type 1 to dextran sulfate associated with the emergence of specific mutations in the envelope gp120 glycoprotein. Mol. Pharmacol. 1997, 52, 98–104. [Google Scholar]
- Moulard, M.; Lortat-Jacob, H.; Mondor, I.; Roca, G.; Wyatt, R.; Sodroski, J.; Zhao, L.; Olson, W.; Kwong, P.D.; Sattentau, Q.J. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 2000, 74, 1948–1960. [Google Scholar]
- Este, J.A.; Cabrera, C.; Blanco, J.; Gutierrez, A.; Bridger, G.; Henson, G.; Clotet, B.; Schols, D.; de Clercq, E. Shift of clinical human immunodeficiency virus type 1 isolates from X4 to R5 and prevention of emergence of the syncytium-inducing phenotype by blockade of CXCR4. J. Virol. 1999, 73, 5577–5585. [Google Scholar]
- Waters, L.; Mandalia, S.; Randell, P.; Wildfire, A.; Gazzard, B.; Moyle, G. The impact of HIV tropism on decreases in CD4 cell count, clinical progression, and subsequent response to a first antiretroviral therapy regimen. Clin. Infect. Dis. 2008, 46, 1617–1623. [Google Scholar] [CrossRef]
- Maeda, Y.; Foda, M.; Matsushita, S.; Harada, S. Involvement of both the V2 and V3 regions of the CCR5-tropic human immunodeficiency virus type 1 envelope in reduced sensitivity to macrophage inflammatory protein 1alpha. J. Virol. 2000, 74, 1787–1793. [Google Scholar] [CrossRef]
- Aarons, E.J.; Beddows, S.; Willingham, T.; Wu, L.; Koup, R.A. Adaptation to blockade of human immunodeficiency virus type 1 entry imposed by the anti-CCR5 monoclonal antibody 2D7. Virology 2001, 287, 382–390. [Google Scholar] [CrossRef]
- Mosier, D.E.; Picchio, G.R.; Gulizia, R.J.; Sabbe, R.; Poignard, P.; Picard, L.; Offord, R.E.; Thompson, D.A.; Wilken, J. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J. Virol. 1999, 73, 3544–3550. [Google Scholar]
- Trkola, A.; Kuhmann, S.E.; Strizki, J.M.; Maxwell, E.; Ketas, T.; Morgan, T.; Pugach, P.; Xu, S.; Wojcik, L.; Tagat, J.; et al. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 395–400. [Google Scholar]
- Marozsan, A.J.; Kuhmann, S.E.; Morgan, T.; Herrera, C.; Rivera-Troche, E.; Xu, S.; Baroudy, B.M.; Strizki, J.; Moore, J.P. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology 2005, 338, 182–199. [Google Scholar] [CrossRef]
- Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 2007, 81, 2359–2371. [Google Scholar] [CrossRef]
- Strizki, J.M.; Tremblay, C.; Xu, S.; Wojcik, L.; Wagner, N.; Gonsiorek, W.; Hipkin, R.W.; Chou, C.C.; Pugliese-Sivo, C.; Xiao, Y.; et al. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2005, 49, 4911–4919. [Google Scholar] [CrossRef]
- Ogert, R.A.; Wojcik, L.; Buontempo, C.; Ba, L.; Buontempo, P.; Ralston, R.; Strizki, J.; Howe, J.A. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology 2008, 373, 387–399. [Google Scholar] [CrossRef]
- Baba, M.; Miyake, H.; Wang, X.; Okamoto, M.; Takashima, K. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob. Agents Chemother. 2007, 51, 707–715. [Google Scholar] [CrossRef]
- Kitrinos, K.M.; Amrine-Madsen, H.; Irlbeck, D.M.; Word, J.M.; Demarest, J.F. Virologic failure in therapy-naive subjects on aplaviroc plus lopinavir-ritonavir: Detection of aplaviroc resistance requires clonal analysis of envelope. Antimicrob. Agents Chemother. 2009, 53, 1124–1131. [Google Scholar] [CrossRef]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar]
- Lalezari, J.; Thompson, M.; Kumar, P.; Piliero, P.; Davey, R.; Patterson, K.; Shachoy-Clark, A.; Adkison, K.; Demarest, J.; Lou, Y.; et al. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS 2005, 19, 1443–1448. [Google Scholar] [CrossRef]
- Fatkenheuer, G.; Pozniak, A.L.; Johnson, M.A.; Plettenberg, A.; Staszewski, S.; Hoepelman, A.I.; Saag, M.S.; Goebel, F.D.; Rockstroh, J.K.; Dezube, B.J.; et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 2005, 11, 1170–1172. [Google Scholar]
- Gulick, R.M.; Su, Z.; Flexner, C.; Hughes, M.D.; Skolnik, P.R.; Wilkin, T.J.; Gross, R.; Krambrink, A.; Coakley, E.; Greaves, W.L.; et al. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-Infected, treatment-experienced patients: AIDS clinical trials group 5211. J. Infect. Dis. 2007, 196, 304–312. [Google Scholar] [CrossRef]
- Schurmann, D.; Fatkenheuer, G.; Reynes, J.; Michelet, C.; Raffi, F.; van Lier, J.; Caceres, M.; Keung, A.; Sansone-Parsons, A.; Dunkle, L.M.; et al. Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults. AIDS 2007, 21, 1293–1299. [Google Scholar] [CrossRef]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. New Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef]
- Landovitz, R.J.; Angel, J.B.; Hoffmann, C.; Horst, H.; Opravil, M.; Long, J.; Greaves, W.; Fatkenheuer, G. Phase II study of vicriviroc versus efavirenz (both with zidovudine/lamivudine) in treatment-naive subjects with HIV-1 infection. J. Infect. Dis. 2008, 198, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Tsibris, A.M.; Sagar, M.; Gulick, R.M.; Su, Z.; Hughes, M.; Greaves, W.; Subramanian, M.; Flexner, C.; Giguel, F.; Leopold, K.E.; et al. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J. Virol. 2008, 82, 8210–8214. [Google Scholar] [CrossRef]
- Yeni, P.; Lamarca, A.; Berger, D.; Cimoch, P.; Lazzarin, A.; Salvato, P.; Smaill, F.M.; Teofilo, E.; Madison, S.J.; Nichols, W.G.; et al. Antiviral activity and safety of aplaviroc, a CCR5 antagonist, in combination with lopinavir/ritonavir in HIV-infected, therapy-naive patients: Results of the EPIC study (CCR100136). HIV Med. 2009, 10, 116–124. [Google Scholar] [CrossRef]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for previously treated patients with R5 HIV-1 infection. New Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar]
- Cooper, D.A.; Heera, J.; Goodrich, J.; Tawadrous, M.; Saag, M.; Dejesus, E.; Clumeck, N.; Walmsley, S.; Ting, N.; Coakley, E.; et al. Maraviroc versus efavirenz, both in combination with zidovudine-lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J. Infect. Dis. 2010, 201, 803–813. [Google Scholar] [CrossRef]
- Baatz, F.; Struck, D.; Lemaire, M.; de Landtsheer, S.; Servais, J.Y.; Arendt, V.; Schmit, J.C.; Perez Bercoff, D. Rescue of HIV-1 long-time archived X4 strains to escape maraviroc. Antivir. Res. 2011, 92, 488–492. [Google Scholar]
- Cilliers, T.; Nhlapo, J.; Coetzer, M.; Orlovic, D.; Ketas, T.; Olson, W.C.; Moore, J.P.; Trkola, A.; Morris, L. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from subtype C. J. Virol. 2003, 77, 4449–4456. [Google Scholar]
- Coetzer, M.; Nedellec, R.; Cilliers, T.; Meyers, T.; Morris, L.; Mosier, D.E. Extreme genetic divergence is required for coreceptor switching in HIV-1 subtype C. J. Acquir. Immune Defic. Syndr. 2011, 56, 9–15. [Google Scholar] [CrossRef]
- Kuhmann, S.E.; Pugach, P.; Kunstman, K.J.; Taylor, J.; Stanfield, R.L.; Snyder, A.; Strizki, J.M.; Riley, J.; Baroudy, B.M.; Wilson, I.A.; et al. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J. Virol. 2004, 78, 2790–2807. [Google Scholar]
- Pugach, P.; Marozsan, A.J.; Ketas, T.J.; Landes, E.L.; Moore, J.P.; Kuhmann, S.E. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology 2007, 361, 212–228. [Google Scholar] [CrossRef]
- Anastassopoulou, C.G.; Ketas, T.J.; Klasse, P.J.; Moore, J.P. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5318–5323. [Google Scholar]
- Berro, R.; Sanders, R.W.; Lu, M.; Klasse, P.J.; Moore, J.P. Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry. PLoS Pathog. 2009, 5, e1000548. [Google Scholar] [CrossRef]
- Nolan, K.M.; Del Prete, G.Q.; Jordan, A.P.; Haggarty, B.; Romano, J.; Leslie, G.J.; Hoxie, J.A. Characterization of a human immunodeficiency virus type 1 V3 deletion mutation that confers resistance to CCR5 inhibitors and the ability to use aplaviroc-bound receptor. J. Virol. 2009, 83, 3798–3809. [Google Scholar] [CrossRef]
- Berro, R.; Klasse, P.J.; Jakobsen, M.R.; Gorry, P.R.; Moore, J.P.; Sanders, R.W. V3 determinants of HIV-1 escape from the CCR5 inhibitors Maraviroc and Vicriviroc. Virology 2012, 427, 158–165. [Google Scholar] [CrossRef]
- Ogert, R.A.; Ba, L.; Hou, Y.; Buontempo, C.; Qiu, P.; Duca, J.; Murgolo, N.; Buontempo, P.; Ralston, R.; Howe, J.A. Structure-function analysis of human immunodeficiency virus type 1 gp120 amino acid mutations associated with resistance to the CCR5 coreceptor antagonist vicriviroc. J. Virol. 2009, 83, 12151–12163. [Google Scholar]
- Yuan, Y.; Maeda, Y.; Terasawa, H.; Monde, K.; Harada, S.; Yusa, K. A combination of polymorphic mutations in V3 loop of HIV-1 gp120 can confer noncompetitive resistance to maraviroc. Virology 2011, 413, 293–299. [Google Scholar]
- Laakso, M.M.; Lee, F.H.; Haggarty, B.; Agrawal, C.; Nolan, K.M.; Biscone, M.; Romano, J.; Jordan, A.P.; Leslie, G.J.; Meissner, E.G.; et al. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 2007, 3, e117. [Google Scholar] [CrossRef]
- Yusa, K.; Maeda, Y.; Fujioka, A.; Monde, K.; Harada, S. Isolation of TAK-779-resistant HIV-1 from an R5 HIV-1 GP120 V3 loop library. J. Biol. Chem. 2005, 280, 30083–30090. [Google Scholar]
- McNicholas, P.M.; Mann, P.A.; Wojcik, L.; Phd, P.Q.; Lee, E.; McCarthy, M.; Shen, J.; Black, T.A.; Strizki, J.M. Mapping and characterization of vicriviroc resistance mutations from HIV-1 isolated from treatment-experienced subjects enrolled in a phase II study (VICTOR-E1). J. Acquir. Immune Defic. Syndr. 2011, 56, 222–229. [Google Scholar] [CrossRef]
- Ogert, R.A.; Hou, Y.; Ba, L.; Wojcik, L.; Qiu, P.; Murgolo, N.; Duca, J.; Dunkle, L.M.; Ralston, R.; Howe, J.A. Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology 2010, 400, 145–155. [Google Scholar] [CrossRef]
- Pfaff, J.M.; Wilen, C.B.; Harrison, J.E.; Demarest, J.F.; Lee, B.; Doms, R.W.; Tilton, J.C. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 2010, 84, 6505–6514. [Google Scholar] [CrossRef]
- McNicholas, P.; Wei, Y.; Whitcomb, J.; Greaves, W.; Black, T.A.; Tremblay, C.L.; Strizki, J.M. Characterization of emergent HIV resistance in treatment-naive subjects enrolled in a vicriviroc phase 2 trial. J. Infect. Dis. 2010, 201, 1470–1480. [Google Scholar] [CrossRef]
- Henrich, T.J.; Tsibris, A.M.; Lewine, N.R.; Konstantinidis, I.; Leopold, K.E.; Sagar, M.; Kuritzkes, D.R. Evolution of CCR5 antagonist resistance in an HIV-1 subtype C clinical isolate. J. Acquir. Immune Defic. Syndr. 2010, 55, 420–427. [Google Scholar] [CrossRef]
- Anastassopoulou, C.G.; Ketas, T.J.; Depetris, R.S.; Thomas, A.M.; Klasse, P.J.; Moore, J.P. Resistance of a human immunodeficiency virus type 1 isolate to a small molecule CCR5 inhibitor can involve sequence changes in both gp120 and gp41. Virology 2011, 413, 47–59. [Google Scholar] [CrossRef]
- Moore, J.P.; Kuritzkes, D.R. A piece de resistance: How HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV AIDS 2009, 4, 118–124. [Google Scholar] [CrossRef]
- Lobritz, M.A.; Ratcliff, A.N.; Arts, E.J. HIV-1 Entry, Inhibitors, and Resistance. Viruses 2010, 2, 1069–1105. [Google Scholar] [CrossRef]
- Tilton, J.C.; Amrine-Madsen, H.; Miamidian, J.L.; Kitrinos, K.M.; Pfaff, J.; Demarest, J.F.; Ray, N.; Jeffrey, J.L.; Labranche, C.C.; Doms, R.W. HIV type 1 from a patient with baseline resistance to CCR5 antagonists uses drug-bound receptor for entry. AIDS Res. Hum. Retrovir. 2010, 26, 13–24. [Google Scholar] [CrossRef]
- Pugach, P.; Ray, N.; Klasse, P.J.; Ketas, T.J.; Michael, E.; Doms, R.W.; Lee, B.; Moore, J.P. Inefficient entry of vicriviroc-resistant HIV-1 via the inhibitor-CCR5 complex at low cell surface CCR5 densities. Virology 2009, 387, 296–302. [Google Scholar] [CrossRef]
- Heredia, A.; Latinovic, O.; Gallo, R.C.; Melikyan, G.; Reitz, M.; Le, N.; Redfield, R.R. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 20476–20481. [Google Scholar]
- Baribaud, F.; Edwards, T.G.; Sharron, M.; Brelot, A.; Heveker, N.; Price, K.; Mortari, F.; Alizon, M.; Tsang, M.; Doms, R.W. Antigenically distinct conformations of CXCR4. J. Virol. 2001, 75, 8957–8967. [Google Scholar] [CrossRef]
- Berro, R.; Klasse, P.J.; Lascano, D.; Flegler, A.; Nagashima, K.A.; Sanders, R.W.; Sakmar, T.P.; Hope, T.J.; Moore, J.P. Multiple CCR5 conformations on the cell surface are used differentially by human immunodeficiency viruses resistant or sensitive to CCR5 inhibitors. J. Virol. 2011, 85, 8227–8240. [Google Scholar]
- Platt, E.J.; Durnin, J.P.; Shinde, U.; Kabat, D. An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for membrane fusion and overcomes diverse entry limitations. J. Mol. Biol. 2007, 374, 64–79. [Google Scholar] [CrossRef]
- Reeves, J.D.; Gallo, S.A.; Ahmad, N.; Miamidian, J.L.; Harvey, P.E.; Sharron, M.; Pohlmann, S.; Sfakianos, J.N.; Derdeyn, C.A.; Blumenthal, R.; et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16249–16254. [Google Scholar]
- Putcharoen, O.; Lee, S.H.; Henrich, T.J.; Hu, Z.; Vanichanan, J.; Coakley, E.; Greaves, W.; Gulick, R.M.; Kuritzkes, D.R.; Tsibris, A.M. HIV-1 clinical isolates resistant to CCR5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J. Virol. 2012, 86, 1119–1128. [Google Scholar]
- Anastassopoulou, C.G.; Ketas, T.J.; Sanders, R.W.; Klasse, P.J.; Moore, J.P. Effects of sequence changes in the HIV-1 gp41 fusion peptide on CCR5 inhibitor resistance. Virology 2012, 428, 86–97. [Google Scholar] [CrossRef]
- Anastassopoulou, C.G.; Marozsan, A.J.; Matet, A.; Snyder, A.D.; Arts, E.J.; Kuhmann, S.E.; Moore, J.P. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 2007, 3, e79. [Google Scholar] [CrossRef]
- Tsibris, A.M.; Korber, B.; Arnaout, R.; Russ, C.; Lo, C.C.; Leitner, T.; Gaschen, B.; Theiler, J.; Paredes, R.; Su, Z.; et al. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 2009, 4, e5683. [Google Scholar]
- Tsibris, A.M.; Hu, Z.; Paredes, R.; Leopold, K.E.; Putcharoen, O.; Schure, A.L.; Mazur, N.; Coakley, E.; Su, Z.; Gulick, R.M.; et al. Vicriviroc resistance decay and relative replicative fitness in HIV-1 clinical isolates under sequential drug selection pressures. J. Virol. 2012, 86, 6416–6426. [Google Scholar]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000, 151, 413–423. [Google Scholar] [CrossRef]
- Herskowitz, I. Functional inactivation of genes by dominant negative mutations. Nature 1987, 329, 219–222. [Google Scholar] [CrossRef]
- Amaya, E.; Musci, T.J.; Kirschner, M.W. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell 1991, 66, 257–270. [Google Scholar] [CrossRef]
- Matthews, T.J.; Wild, C.; Chen, C.H.; Bolognesi, D.P.; Greenberg, M.L. Structural rearrangements in the transmembrane glycoprotein after receptor binding. Immunol. Rev. 1994, 140, 93–104. [Google Scholar] [CrossRef]
- Highlights of prescribing information. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021481s025lbl.pdf (accessed on 6 November 2012).
- Lalezari, J.P.; Henry, K.; O'Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. New Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef]
- Kilby, J.M.; Eron, J.J. Novel therapies based on mechanisms of HIV-1 cell entry. New Engl. J. Med. 2003, 348, 2228–2238. [Google Scholar] [CrossRef]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 9770–9774. [Google Scholar]
- Wild, C.; Oas, T.; McDanal, C.; Bolognesi, D.; Matthews, T. A synthetic peptide inhibitor of human immunodeficiency virus replication: Correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 10537–10541. [Google Scholar]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar]
- Wild, C.; Greenwell, T.; Shugars, D.; Rimsky-Clarke, L.; Matthews, T. The inhibitory activity of an HIV type 1 peptide correlates with its ability to interact with a leucine zipper structure. AIDS Res. Hum. Retrovir. 1995, 11, 323–325. [Google Scholar] [CrossRef]
- Chen, C.H.; Matthews, T.J.; McDanal, C.B.; Bolognesi, D.P.; Greenberg, M.L. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: Implication for viral fusion. J. Virol. 1995, 69, 3771–3777. [Google Scholar]
- Tan, K.; Liu, J.; Wang, J.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12303–12308. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Liu, S.; Jing, W.; Cheung, B.; Lu, H.; Sun, J.; Yan, X.; Niu, J.; Farmar, J.; Wu, S.; Jiang, S. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J. Biol. Chem. 2007, 282, 9612–9620. [Google Scholar]
- Kliger, Y.; Gallo, S.A.; Peisajovich, S.G.; Munoz-Barroso, I.; Avkin, S.; Blumenthal, R.; Shai, Y. Mode of action of an antiviral peptide from HIV-1. Inhibition at a post-lipid mixing stage. J. Biol. Chem. 2001, 276, 1391–1397. [Google Scholar] [CrossRef]
- Yuan, W.; Craig, S.; Si, Z.; Farzan, M.; Sodroski, J. CD4-induced T-20 binding to human immunodeficiency virus type 1 gp120 blocks interaction with the CXCR4 coreceptor. J. Virol. 2004, 78, 5448–5457. [Google Scholar] [CrossRef]
- Alam, S.M.; Paleos, C.A.; Liao, H.X.; Scearce, R.; Robinson, J.; Haynes, B.F. An inducible HIV type 1 gp41 HR-2 peptide-binding site on HIV type 1 envelope gp120. AIDS Res. Hum. Retrovir. 2004, 20, 836–845. [Google Scholar] [CrossRef]
- He, Y.; Vassell, R.; Zaitseva, M.; Nguyen, N.; Yang, Z.; Weng, Y.; Weiss, C.D. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J. Virol. 2003, 77, 1666–1671. [Google Scholar] [CrossRef]
- Furuta, R.A.; Wild, C.T.; Weng, Y.; Weiss, C.D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998, 5, 276–279. [Google Scholar] [CrossRef]
- Koshiba, T.; Chan, D.C. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J. Biol. Chem. 2003, 278, 7573–7579. [Google Scholar] [CrossRef]
- Kilgore, N.R.; Salzwedel, K.; Reddick, M.; Allaway, G.P.; Wild, C.T. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J. Virol. 2003, 77, 7669–7672. [Google Scholar]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef]
- Poveda, E.; Rodes, B.; Toro, C.; Martin-Carbonero, L.; Gonzalez-Lahoz, J.; Soriano, V. Evolution of the gp41 env region in HIV-infected patients receiving T-20, a fusion inhibitor. AIDS 2002, 16, 1959–1961. [Google Scholar] [CrossRef]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar]
- Nameki, D.; Kodama, E.; Ikeuchi, M.; Mabuchi, N.; Otaka, A.; Tamamura, H.; Ohno, M.; Fujii, N.; Matsuoka, M. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J. Virol. 2005, 79, 764–770. [Google Scholar]
- Poveda, E.; Rodes, B.; Lebel-Binay, S.; Faudon, J.L.; Jimenez, V.; Soriano, V. Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J. Clin. Virol. 2005, 34, 295–301. [Google Scholar] [CrossRef]
- Perez-Alvarez, L.; Carmona, R.; Ocampo, A.; Asorey, A.; Miralles, C.; Perez de Castro, S.; Pinilla, M.; Contreras, G.; Taboada, J.A.; Najera, R. Long-term monitoring of genotypic and phenotypic resistance to T20 in treated patients infected with HIV-1. J. Med. Virol. 2006, 78, 141–147. [Google Scholar] [CrossRef]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar]
- Cilliers, T.; Moore, P.; Coetzer, M.; Morris, L. In vitro generation of HIV type 1 subtype C isolates resistant to enfuvirtide. AIDS Res. Hum. Retrovir. 2005, 21, 776–783. [Google Scholar] [CrossRef]
- Cabrera, C.; Marfil, S.; Garcia, E.; Martinez-Picado, J.; Bonjoch, A.; Bofill, M.; Moreno, S.; Ribera, E.; Domingo, P.; Clotet, B.; et al. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. AIDS 2006, 20, 2075–2080. [Google Scholar]
- Menzo, S.; Castagna, A.; Monachetti, A.; Hasson, H.; Danise, A.; Carini, E.; Bagnarelli, P.; Lazzarin, A.; Clementi, M. Genotype and phenotype patterns of human immunodeficiency virus type 1 resistance to enfuvirtide during long-term treatment. Antimicrob. Agents Chemother. 2004, 48, 3253–3259. [Google Scholar] [CrossRef]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar]
- Lu, J.; Deeks, S.G.; Hoh, R.; Beatty, G.; Kuritzkes, B.A.; Martin, J.N.; Kuritzkes, D.R. Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: Results of a clonal analysis. J. Acquir. Immune Defic. Syndr. 2006, 43, 60–64. [Google Scholar] [CrossRef]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar]
- Trivedi, V.D.; Cheng, S.F.; Wu, C.W.; Karthikeyan, R.; Chen, C.J.; Chang, D.K. The LLSGIV stretch of the N-terminal region of HIV-1 gp41 is critical for binding to a model peptide, T20. Protein Eng. 2003, 16, 311–317. [Google Scholar] [CrossRef]
- Hanna, S.L.; Yang, C.; Owen, S.M.; Lal, R.B. Variability of critical epitopes within HIV-1 heptad repeat domains for selected entry inhibitors in HIV-infected populations worldwide [corrected]. AIDS 2002, 16, 1603–1608. [Google Scholar] [CrossRef]
- Labrosse, B.; Labernardiere, J.L.; Dam, E.; Trouplin, V.; Skrabal, K.; Clavel, F.; Mammano, F. Baseline susceptibility of primary human immunodeficiency virus type 1 to entry inhibitors. J. Virol. 2003, 77, 1610–1613. [Google Scholar] [CrossRef]
- Xu, L.; Hue, S.; Taylor, S.; Ratcliffe, D.; Workman, J.A.; Jackson, S.; Cane, P.A.; Pillay, D. Minimal variation in T-20 binding domain of different HIV-1 subtypes from antiretroviral-naive and -experienced patients. AIDS 2002, 16, 1684–1686. [Google Scholar] [CrossRef]
- Zollner, B.; Feucht, H.H.; Schroter, M.; Schafer, P.; Plettenberg, A.; Stoehr, A.; Laufs, R. Primary genotypic resistance of HIV-1 to the fusion inhibitor T-20 in long-term infected patients. AIDS 2001, 15, 935–936. [Google Scholar] [CrossRef]
- Roman, F.; Gonzalez, D.; Lambert, C.; Deroo, S.; Fischer, A.; Baurith, T.; Staub, T.; Boulme, R.; Arendt, V.; Schneider, F.; et al. Uncommon mutations at residue positions critical for enfuvirtide (T-20) resistance in enfuvirtide-naive patients infected with subtype B and non-B HIV-1 strains. J. Acquir. Immune Defic. Syndr. 2003, 33, 134–139. [Google Scholar] [CrossRef]
- Villahermosa, M.L.; Perez-Alvarez, L.; Carmona, R.; Cuevas, M.T.; Thomson, M.M.; Medrano, L.; Vazquez de Parga, E.; Delgado, E.; Pedreira, J.D.; Najera, R. Primary resistance mutations to fusion inhibitors and polymorphisms in gp41 sequences of HIV-1 non-B subtypes and recombinants. AIDS 2003, 17, 1083–1086. [Google Scholar] [CrossRef]
- Loutfy, M.R.; Raboud, J.M.; Montaner, J.S.; Antoniou, T.; Wynhoven, B.; Smaill, F.; Rouleau, D.; Gill, J.; Schlech, W.; Brumme, Z.L.; et al. Assay of HIV gp41 amino acid sequence to identify baseline variation and mutation development in patients with virologic failure on enfuvirtide. Antivir. Res. 2007, 75, 58–63. [Google Scholar]
- Caffrey, M. Model for the structure of the HIV gp41 ectodomain: Insight into the intermolecular interactions of the gp41 loop. Biochim. Biophys. Acta 2007, 1536, 116–122. [Google Scholar]
- Caffrey, M.; Cai, M.; Kaufman, J.; Stahl, S.J.; Wingfield, P.T.; Covell, D.G.; Gronenborn, A.M.; Clore, G.M. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998, 17, 4572–4584. [Google Scholar] [CrossRef]
- Melby, T.; Sista, P.; DeMasi, R.; Kirkland, T.; Roberts, N.; Salgo, M.; Heilek-Snyder, G.; Cammack, N.; Matthews, T.J.; Greenberg, M.L. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Res. Hum. Retrovir. 2006, 22, 375–385. [Google Scholar] [CrossRef]
- Tolstrup, M.; Selzer-Plon, J.; Laursen, A.L.; Bertelsen, L.; Gerstoft, J.; Duch, M.; Pedersen, F.S.; Ostergaard, L. Full fusion competence rescue of the enfuvirtide resistant HIV-1 gp41 genotype (43D) by a prevalent polymorphism (137K). AIDS 2007, 21, 519–521. [Google Scholar] [CrossRef]
- Ray, N.; Blackburn, L.A.; Doms, R.W. HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J. Virol. 2009, 83, 2989–2995. [Google Scholar] [CrossRef]
- Bai, X.; Wilson, K.L.; Seedorff, J.E.; Ahrens, D.; Green, J.; Davison, D.K.; Jin, L.; Stanfield-Oakley, S.A.; Mosier, S.M.; Melby, T.E.; et al. Impact of the enfuvirtide resistance mutation N43D and the associated baseline polymorphism E137K on peptide sensitivity and six-helix bundle structure. Biochemistry 2008, 47, 6662–6670. [Google Scholar]
- Baatz, F.; Nijhuis, M.; Lemaire, M.; Riedijk, M.; Wensing, A.M.; Servais, J.Y.; van Ham, P.M.; Hoepelman, A.I.; Koopmans, P.P.; Sprenger, H.G.; et al. Impact of the HIV-1 env genetic context outside HR1-HR2 on resistance to the fusion inhibitor enfuvirtide and viral infectivity in clinical isolates. PLoS One 2011, 6, e21535. [Google Scholar]
- Holguin, A.; Faudon, J.L.; Labernardiere, J.L.; Soriano, V. Susceptibility of HIV-1 non-B subtypes and recombinant variants to Enfuvirtide. J. Clin. Virol. 2007, 38, 176–180. [Google Scholar] [CrossRef]
- Labrosse, B.; Morand-Joubert, L.; Goubard, A.; Rochas, S.; Labernardiere, J.L.; Pacanowski, J.; Meynard, J.L.; Hance, A.J.; Clavel, F.; Mammano, F. Role of the envelope genetic context in the development of enfuvirtide resistance in human immunodeficiency virus type 1-infected patients. J. Virol. 2006, 80, 8807–8819. [Google Scholar]
- Goubard, A.; Clavel, F.; Mammano, F.; Labrosse, B. In vivo selection by enfuvirtide of HIV type-1 env quasispecies with optimal potential for phenotypic expression of HR1 mutations. Antivir. Ther. 2009, 14, 597–602. [Google Scholar]
- Lu, J.; Sista, P.; Giguel, F.; Greenberg, M.; Kuritzkes, D.R. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 2004, 78, 4628–4637. [Google Scholar] [CrossRef]
- Marconi, V.; Bonhoeffer, S.; Paredes, R.; Lu, J.; Hoh, R.; Martin, J.N.; Deeks, S.G.; Kuritzkes, D.R. Viral dynamics and in vivo fitness of HIV-1 in the presence and absence of enfuvirtide. J. Acquir. Immune Defic. Syndr. 2008, 48, 572–576. [Google Scholar] [CrossRef]
- Watabe, T.; Terakawa, Y.; Watanabe, K.; Ohno, H.; Nakano, H.; Nakatsu, T.; Kato, H.; Izumi, K.; Kodama, E.; Matsuoka, M.; et al. X-ray crystallographic study of an HIV-1 fusion inhibitor with the gp41 S138A substitution. J. Mol. Biol. 2009, 392, 657–665. [Google Scholar] [CrossRef]
- Baldwin, C.; Berkhout, B. Mechanistic studies of a T20-dependent human immunodeficiency virus type 1 variant. J. Virol. 2008, 82, 7735–7740. [Google Scholar]
- Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 15613–15617. [Google Scholar]
- Otaka, A.; Nakamura, M.; Nameki, D.; Kodama, E.; Uchiyama, S.; Nakamura, S.; Nakano, H.; Tamamura, H.; Kobayashi, Y.; Matsuoka, M.; et al. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew. Chem. 2002, 41, 2937–2940. [Google Scholar] [CrossRef]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Zhang, Z.; O'Brien, W.A.; Ratner, L.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J. Virol. 2001, 75, 8605–8614. [Google Scholar]
- He, Y.; Cheng, J.; Li, J.; Qi, Z.; Lu, H.; Dong, M.; Jiang, S.; Dai, Q. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: Implications for designing novel anti-HIV fusion inhibitors. J. Virol. 2008, 82, 6349–6358. [Google Scholar] [CrossRef]
- Greenberg, M.L.; Davison, D.; Jin, L.; Mosier, S.; Melby, T.; Sista, P.; Demasi, R.; Miralles, D.; Cammack, N.; Matthews, T.J. In vitro antiviral activity of T-1249, a second generation fusion inhibitor. Antivir. Ther. 2002, 7, S10. [Google Scholar]
- Eron, J.J.; Gulick, R.M.; Bartlett, J.A.; Merigan, T.; Arduino, R.; Kilby, J.M.; Yangco, B.; Diers, A.; Drobnes, C.; DeMasi, R.; et al. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J. Infect. Dis. 2004, 189, 1075–1083. [Google Scholar] [CrossRef]
- Chong, H.; Yao, X.; Sun, J.; Qiu, Z.; Zhang, M.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. The M-T Hook Structure Is Critical for Design of HIV-1 Fusion Inhibitors. J. Biol. Chem. 2012, 287, 34558–34568. [Google Scholar]
- Chong, H.; Yao, X.; Qiu, Z.; Qin, B.; Han, R.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Discovery of critical residues for viral entry and inhibition through structural Insight of HIV-1 fusion inhibitor CP621–652. J. Biol. Chem. 2012, 287, 20281–20289. [Google Scholar]
- Melby, T.; Demasi, R.; Cammack, N.; Miralles, G.D.; Greenberg, M.L. Evolution of genotypic and phenotypic resistance during chronic treatment with the fusion inhibitor T-1249. AIDS Res. Hum. Retrovir. 2007, 23, 1366–1373. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Rajan, D.; Munch, J.; Kirchhoff, F. Human immunodeficiency virus type 1 variants resistant to first- and second-version fusion inhibitors and cytopathic in ex vivo human lymphoid tissue. J. Virol. 2007, 81, 6563–6572. [Google Scholar] [CrossRef]
- Eggink, D.; Baldwin, C.E.; Deng, Y.; Langedijk, J.P.; Lu, M.; Sanders, R.W.; Berkhout, B. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J. Virol. 2008, 82, 6678–6688. [Google Scholar]
- Lalezari, J.P.; Bellos, N.C.; Sathasivam, K.; Richmond, G.J.; Cohen, C.J.; Myers, R.A., Jr.; Henry, D.H.; Raskino, C.; Melby, T.; Murchison, H.; et al. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J. Infect. Dis. 2005, 191, 1155–1163. [Google Scholar] [CrossRef]
- Armand-Ugon, M.; Gutierrez, A.; Clotet, B.; Este, J.A. HIV-1 resistance to the gp41-dependent fusion inhibitor C-34. Antivir. Res. 2003, 59, 137–142. [Google Scholar]
- Eggink, D.; Langedijk, J.P.; Bonvin, A.M.; Deng, Y.; Lu, M.; Berkhout, B.; Sanders, R.W. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J. Biol. Chem. 2009, 284, 26941–26950. [Google Scholar]
- Naito, T.; Izumi, K.; Kodama, E.; Sakagami, Y.; Kajiwara, K.; Nishikawa, H.; Watanabe, K.; Sarafianos, S.G.; Oishi, S.; Fujii, N.; et al. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob. Agents Chemother. 2009, 53, 1013–1018. [Google Scholar] [CrossRef]
- Nishikawa, H.; Nakamura, S.; Kodama, E.; Ito, S.; Kajiwara, K.; Izumi, K.; Sakagami, Y.; Oishi, S.; Ohkubo, T.; Kobayashi, Y.; et al. Electrostatically constrained alpha-helical peptide inhibits replication of HIV-1 resistant to enfuvirtide. Int. J. Biochem. Cell Biol. 2009, 41, 891–899. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cheng, J.; Lu, H.; Li, J.; Hu, J.; Qi, Z.; Liu, Z.; Jiang, S.; Dai, Q. Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16332–16337. [Google Scholar]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar]
- Yao, X.; Chong, H.; Zhang, C.; Qiu, Z.; Qin, B.; Han, R.; Waltersperger, S.; Wang, M.; He, Y.; Cui, S. Structural basis of potent and broad HIV-1 fusion inhibitor CP32M. J. Biol. Chem. 2012, 287, 26618–26629. [Google Scholar]
- Yao, X.; Chong, H.; Zhang, C.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J. Biol. Chem. 2012, 287, 6788–6796. [Google Scholar]
- Yu, X.; Lu, L.; Cai, L.; Tong, P.; Tan, S.; Zou, P.; Meng, F.; Chen, Y.H.; Jiang, S. Mutations of Gln64 in the HIV-1 gp41 N-terminal heptad repeat render viruses resistant to peptide HIV fusion inhibitors targeting the gp41 pocket. J. Virol. 2012, 86, 589–593. [Google Scholar]
- Liu, Z.; Shan, M.; Li, L.; Lu, L.; Meng, S.; Chen, C.; He, Y.; Jiang, S.; Zhang, L. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2011, 286, 3277–3287. [Google Scholar]
- Shimura, K.; Nameki, D.; Kajiwara, K.; Watanabe, K.; Sakagami, Y.; Oishi, S.; Fujii, N.; Matsuoka, M.; Sarafianos, S.G.; Kodama, E.N. Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J. Biol. Chem. 2010, 285, 39471–39480. [Google Scholar]
- Izumi, K.; Kodama, E.; Shimura, K.; Sakagami, Y.; Watanabe, K.; Ito, S.; Watabe, T.; Terakawa, Y.; Nishikawa, H.; Sarafianos, S.G.; et al. Design of peptide-based inhibitors for human immunodeficiency virus type 1 strains resistant to T-20. J. Biol. Chem. 2009, 284, 4914–4920. [Google Scholar]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12772–12777. [Google Scholar]
- Kahle, K.M.; Steger, H.K.; Root, M.J. Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 2009, 5, e1000674. [Google Scholar] [CrossRef]
- Gallo, S.A.; Sackett, K.; Rawat, S.S.; Shai, Y.; Blumenthal, R. The stability of the intact envelope glycoproteins is a major determinant of sensitivity of HIV/SIV to peptidic fusion inhibitors. J. Mol. Biol. 2004, 340, 9–14. [Google Scholar]
- Eggink, D.; Bontjer, I.; Langedijk, J.P.; Berkhout, B.; Sanders, R.W. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J. Virol. 2011, 85, 10785–10797. [Google Scholar] [CrossRef]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5801–5806. [Google Scholar]
- Hildinger, M.; Dittmar, M.T.; Schult-Dietrich, P.; Fehse, B.; Schnierle, B.S.; Thaler, S.; Stiegler, G.; Welker, R.; von Laer, D. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J. Virol. 2001, 75, 3038–3042. [Google Scholar] [CrossRef]
- Egelhofer, M.; Brandenburg, G.; Martinius, H.; Schult-Dietrich, P.; Melikyan, G.; Kunert, R.; Baum, C.; Choi, I.; Alexandrov, A.; von Laer, D. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J. Virol. 2004, 78, 568–575. [Google Scholar] [CrossRef]
- Peisajovich, S.G.; Gallo, S.A.; Blumenthal, R.; Shai, Y. C-terminal octylation rescues an inactive T20 mutant: Implications for the mechanism of HIV/SIMIAN immunodeficiency virus-induced membrane fusion. J. Biol. Chem. 2003, 278, 21012–21017. [Google Scholar]
- Hermann, F.G.; Egerer, L.; Brauer, F.; Gerum, C.; Schwalbe, H.; Dietrich, U.; von Laer, D. Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J. Virol. 2009, 83, 4844–4853. [Google Scholar] [CrossRef]
- Lohrengel, S.; Hermann, F.; Hagmann, I.; Oberwinkler, H.; Scrivano, L.; Hoffmann, C.; von Laer, D.; Dittmar, M.T. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J. Virol. 2005, 79, 10237–10246. [Google Scholar] [CrossRef]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16828–16833. [Google Scholar]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O'Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar]
- Reeves, J.D.; Miamidian, J.L.; Biscone, M.J.; Lee, F.H.; Ahmad, N.; Pierson, T.C.; Doms, R.W. Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and entry inhibitor sensitivity. J. Virol. 2004, 78, 5476–5485. [Google Scholar]
- Reeves, J.D.; Lee, F.H.; Miamidian, J.L.; Jabara, C.B.; Juntilla, M.M.; Doms, R.W. Enfuvirtide resistance mutations: Impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 2005, 79, 4991–4999. [Google Scholar]
- Miyauchi, K.; Kozlov, M.M.; Melikyan, G.B. Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 2009, 5, e1000585. [Google Scholar] [CrossRef]
- Abrahamyan, L.G.; Mkrtchyan, S.R.; Binley, J.; Lu, M.; Melikyan, G.B.; Cohen, F.S. The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J. Virol. 2005, 79, 106–115. [Google Scholar]
- Wild, C.; Dubay, J.W.; Greenwell, T.; Baird, T., Jr.; Oas, T.G.; McDanal, C.; Hunter, E.; Matthews, T. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 12676–12680. [Google Scholar]
- Lawless, M.K.; Barney, S.; Guthrie, K.I.; Bucy, T.B.; Petteway, S.R., Jr.; Merutka, G. HIV-1 membrane fusion mechanism: Structural studies of the interactions between biologically-active peptides from gp41. Biochemistry 1996, 35, 13697–13708. [Google Scholar]
- Dwyer, J.J.; Hasan, A.; Wilson, K.L.; White, J.M.; Matthews, T.J.; Delmedico, M.K. The hydrophobic pocket contributes to the structural stability of the N-terminal coiled coil of HIV gp41 but is not required for six-helix bundle formation. Biochemistry 2003, 42, 4945–4953. [Google Scholar] [CrossRef]
- Blacklow, S.C.; Lu, M.; Kim, P.S. A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry 1995, 34, 14955–14962. [Google Scholar] [CrossRef]
- Lu, M.; Kim, P.S. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dynam. 1997, 15, 465–471. [Google Scholar] [CrossRef]
- Eckert, D.M.; Kim, P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11187–11192. [Google Scholar] [CrossRef]
- Louis, J.M.; Bewley, C.A.; Clore, G.M. Design and properties of N(CCG)-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J. Biol. Chem. 2001, 276, 29485–29489. [Google Scholar] [CrossRef]
- Louis, J.M.; Nesheiwat, I.; Chang, L.; Clore, G.M.; Bewley, C.A. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J. Biol. Chem. 2003, 278, 20278–20285. [Google Scholar]
- Chen, X.; Lu, L.; Qi, Z.; Lu, H.; Wang, J.; Yu, X.; Chen, Y.; Jiang, S. Novel recombinant engineered gp41 N-terminal heptad repeat trimers and their potential as anti-HIV-1 therapeutics or microbicides. J. Biol. Chem. 2010, 285, 25506–25515. [Google Scholar]
- Bianchi, E.; Finotto, M.; Ingallinella, P.; Hrin, R.; Carella, A.V.; Hou, X.S.; Schleif, W.A.; Miller, M.D.; Geleziunas, R.; Pessi, A. Covalent stabilization of coiled coils of the HIV gp41 N region yields extremely potent and broad inhibitors of viral infection. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12903–12908. [Google Scholar]
- Wexler-Cohen, Y.; Shai, Y. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog. 2009, 5, e1000509. [Google Scholar] [CrossRef]
- Wexler-Cohen, Y.; Ashkenazi, A.; Viard, M.; Blumenthal, R.; Shai, Y. Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J. 2010, 24, 4196–4202. [Google Scholar] [CrossRef]
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar] [CrossRef]
- Bewley, C.A.; Louis, J.M.; Ghirlando, R.; Clore, G.M. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem. 2002, 277, 14238–14245. [Google Scholar]
- Desmezieres, E.; Gupta, N.; Vassell, R.; He, Y.; Peden, K.; Sirota, L.; Yang, Z.; Wingfield, P.; Weiss, C.D. Human immunodeficiency virus (HIV) gp41 escape mutants: Cross-resistance to peptide inhibitors of HIV fusion and altered receptor activation of gp120. J. Virol. 2005, 79, 4774–4781. [Google Scholar]
- Wang, W.; de Feo, C.J.; Zhuang, M.; Vassell, R.; Weiss, C.D. Selection with a peptide fusion inhibitor corresponding to the first heptad repeat of HIV-1 gp41 identifies two genetic pathways conferring cross-resistance to peptide fusion inhibitors corresponding to the first and second heptad repeats (HR1 and HR2) of gp41. J. Virol. 2011, 85, 12929–12938. [Google Scholar] [CrossRef]
- Zhuang, M.; Wang, W.; de Feo, C.J.; Vassell, R.; Weiss, C.D. Trimeric, coiled-coil extension on peptide fusion inhibitor of HIV-1 influences selection of resistance pathways. J. Biol. Chem. 2012, 287, 8297–8309. [Google Scholar]
- Izumi, K.; Nakamura, S.; Nakano, H.; Shimura, K.; Sakagami, Y.; Oishi, S.; Uchiyama, S.; Ohkubo, T.; Kobayashi, Y.; Fujii, N.; et al. Characterization of HIV-1 resistance to a fusion inhibitor, N36, derived from the gp41 amino-terminal heptad repeat. Antivir. Res. 2010, 87, 179–186. [Google Scholar]
- Steger, H.K.; Root, M.J. Kinetic dependence to HIV-1 entry inhibition. J. Biol. Chem. 2006, 281, 25813–25821. [Google Scholar] [CrossRef]
- O'Brien, W.A.; Sumner-Smith, M.; Mao, S.H.; Sadeghi, S.; Zhao, J.Q.; Chen, I.S. Anti-human immunodeficiency virus type 1 activity of an oligocationic compound mediated via gp120 V3 interactions. J. Virol. 1996, 70, 2825–2831. [Google Scholar]
- Pleskoff, O.; Sol, N.; Marrakchi, H.; Serlin, M.; Seman, M.; Alizon, M. Possible role of the V3 domain of gp120 in resistance to an amphotericin B derivative (MS8209) blocking human immunodeficiency virus entry. J. Virol. 1996, 70, 8247–8251. [Google Scholar]
- Este, J.A.; Cabrera, C.; Schols, D.; Cherepanov, P.; Gutierrez, A.; Witvrouw, M.; Pannecouque, C.; Debyser, Z.; Rando, R.F.; Clotet, B.; et al. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol. Pharmacol. 1998, 53, 340–345. [Google Scholar]
- Munch, J.; Standker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Pohlmann, S.; Chaipan, C.; Biet, T.; Peters, T.; et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef]
- Gonzalez-Ortega, E.; Ballana, E.; Badia, R.; Clotet, B.; Este, J.A. Compensatory mutations rescue the virus replicative capacity of VIRIP-resistant HIV-1. Antivir. Res. 2011, 92, 479–483. [Google Scholar]
- Gonzalez, E.; Ballana, E.; Clotet, B.; Este, J.A. Development of resistance to VIR-353 with cross-resistance to the natural HIV-1 entry virus inhibitory peptide (VIRIP). AIDS 2011, 25, 1557–1583. [Google Scholar] [CrossRef]
- Mayaux, J.F.; Bousseau, A.; Pauwels, R.; Huet, T.; Henin, Y.; Dereu, N.; Evers, M.; Soler, F.; Poujade, C.; de Clercq, E.; et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 3564–3568. [Google Scholar]
- Labrosse, B.; Treboute, C.; Alizon, M. Sensitivity to a nonpeptidic compound (RPR103611) blocking human immunodeficiency virus type 1 Env-mediated fusion depends on sequence and accessibility of the gp41 loop region. J. Virol. 2000, 74, 2142–2150. [Google Scholar] [CrossRef]
- Labrosse, B.; Pleskoff, O.; Sol, N.; Jones, C.; Henin, Y.; Alizon, M. Resistance to a drug blocking human immunodeficiency virus type 1 entry (RPR103611) is conferred by mutations in gp41. J. Virol. 1997, 71, 8230–8236. [Google Scholar]
- Holz-Smith, S.L.; Sun, I.C.; Jin, L.; Matthews, T.J.; Lee, K.H.; Chen, C.H. Role of human immunodeficiency virus (HIV) type 1 envelope in the anti-HIV activity of the betulinic acid derivative IC9564. Antimicrob. Agents Chemother. 2001, 45, 60–66. [Google Scholar] [CrossRef]
- Yuan, X.; Huang, L.; Ho, P.; Labranche, C.; Chen, C.H. Conformation of gp120 determines the sensitivity of HIV-1 DH012 to the entry inhibitor IC9564. Virology 2004, 324, 525–530. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Ho, P.; Li, Z.; Montefiori, D.; Chen, C.H. Betulinic acid derivatives that target gp120 and inhibit multiple genetic subtypes of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2008, 52, 128–136. [Google Scholar] [CrossRef]
- Huang, L.; Lai, W.; Ho, P.; Chen, C.H. Induction of a nonproductive conformational change in gp120 by a small molecule HIV type 1 entry inhibitor. AIDS Res. Hum. Retrovir. 2007, 23, 28–32. [Google Scholar] [CrossRef]
- Murray, E.J.; Leaman, D.P.; Pawa, N.; Perkins, H.; Pickford, C.; Perros, M.; Zwick, M.B.; Butler, S.L. A low-molecular-weight entry inhibitor of both CCR5- and CXCR4-tropic strains of human immunodeficiency virus type 1 targets a novel site on gp41. J. Virol. 2010, 84, 7288–7299. [Google Scholar] [CrossRef]
- Schulz, T.F.; Jameson, B.A.; Lopalco, L.; Siccardi, A.G.; Weiss, R.A.; Moore, J.P. Conserved structural features in the interaction between retroviral surface and transmembrane glycoproteins? AIDS Res. Hum. Retrovir. 1992, 8, 1571–1580. [Google Scholar] [CrossRef]
- Cao, J.; Bergeron, L.; Helseth, E.; Thali, M.; Repke, H.; Sodroski, J. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 1993, 67, 2747–2755. [Google Scholar]
- Merat, R.; Raoul, H.; Leste-Lasserre, T.; Sonigo, P.; Pancino, G. Variable constraints on the principal immunodominant domain of the transmembrane glycoprotein of human immunodeficiency virus type 1. J. Virol. 1999, 73, 5698–5706. [Google Scholar]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar]
- Maerz, A.L.; Drummer, H.E.; Wilson, K.A.; Poumbourios, P. Functional analysis of the disulfide-bonded loop/chain reversal region of human immunodeficiency virus type 1 gp41 reveals a critical role in gp120-gp41 association. J. Virol. 2001, 75, 6635–6644. [Google Scholar] [CrossRef]
- Jacobs, A.; Sen, J.; Rong, L.; Caffrey, M. Alanine scanning mutants of the HIV gp41 loop. J. Biol. Chem. 2005, 280, 27284–27288. [Google Scholar] [CrossRef]
- Sen, J.; Jacobs, A.; Jiang, H.; Rong, L.; Caffrey, M. The disulfide loop of gp41 is critical to the furin recognition site of HIV gp160. Protein Sci. 2007, 16, 1236–1241. [Google Scholar] [CrossRef]
- Kim, S.; Pang, H.B.; Kay, M.S. Peptide mimic of the HIV envelope gp120-gp41 interface. J. Mol. Biol. 2008, 376, 786–797. [Google Scholar]
- Bellamy-McIntyre, A.K.; Bar, S.; Ludlow, L.; Drummer, H.E.; Poumbourios, P. Role for the disulfide-bonded region of human immunodeficiency virus type 1 gp41 in receptor-triggered activation of membrane fusion function. Biochem. Biophys. Res. Comm. 2010, 394, 904–908. [Google Scholar] [CrossRef]
- Finzi, A.; Xiang, S.H.; Pacheco, B.; Wang, L.; Haight, J.; Kassa, A.; Danek, B.; Pancera, M.; Kwong, P.D.; Sodroski, J. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 2010, 37, 656–667. [Google Scholar] [CrossRef]
- Platt, E.J.; Durnin, J.P.; Kabat, D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 2005, 79, 4347–4356. [Google Scholar]
- Mkrtchyan, S.R.; Markosyan, R.M.; Eadon, M.T.; Moore, J.P.; Melikyan, G.B.; Cohen, F.S. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured as an intermediate of membrane fusion. J. Virol. 2005, 79, 11161–11169. [Google Scholar] [CrossRef]
- Abrahamyan, L.G.; Markosyan, R.M.; Moore, J.P.; Cohen, F.S.; Melikyan, G.B. Human immunodeficiency virus type 1 Env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J. Virol. 2003, 77, 5829–5836. [Google Scholar]
- Haim, H.; Strack, B.; Kassa, A.; Madani, N.; Wang, L.; Courter, J.R.; Princiotto, A.; McGee, K.; Pacheco, B.; Seaman, M.S.; et al. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog. 2011, 7, e1002101. [Google Scholar] [CrossRef]
- Xiang, S.H.; Finzi, A.; Pacheco, B.; Alexander, K.; Yuan, W.; Rizzuto, C.; Huang, C.C.; Kwong, P.D.; Sodroski, J. A V3 loop-dependent gp120 element disrupted by CD4 binding stabilizes the human immunodeficiency virus envelope glycoprotein trimer. J. Virol. 2010, 84, 3147–3161. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, L.; Gu, C.; Herschhorn, A.; Xiang, S.H.; Haim, H.; Yang, X.; Sodroski, J. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 2012, 19, 893–899. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Feo, C.J.; Weiss, C.D. Escape from Human Immunodeficiency Virus Type 1 (HIV-1) Entry Inhibitors. Viruses 2012, 4, 3859-3911. https://doi.org/10.3390/v4123859
De Feo CJ, Weiss CD. Escape from Human Immunodeficiency Virus Type 1 (HIV-1) Entry Inhibitors. Viruses. 2012; 4(12):3859-3911. https://doi.org/10.3390/v4123859
Chicago/Turabian StyleDe Feo, Christopher J., and Carol D. Weiss. 2012. "Escape from Human Immunodeficiency Virus Type 1 (HIV-1) Entry Inhibitors" Viruses 4, no. 12: 3859-3911. https://doi.org/10.3390/v4123859
APA StyleDe Feo, C. J., & Weiss, C. D. (2012). Escape from Human Immunodeficiency Virus Type 1 (HIV-1) Entry Inhibitors. Viruses, 4(12), 3859-3911. https://doi.org/10.3390/v4123859