Poxvirus Cell Entry: How Many Proteins Does it Take?

Abstract
:1. Introduction
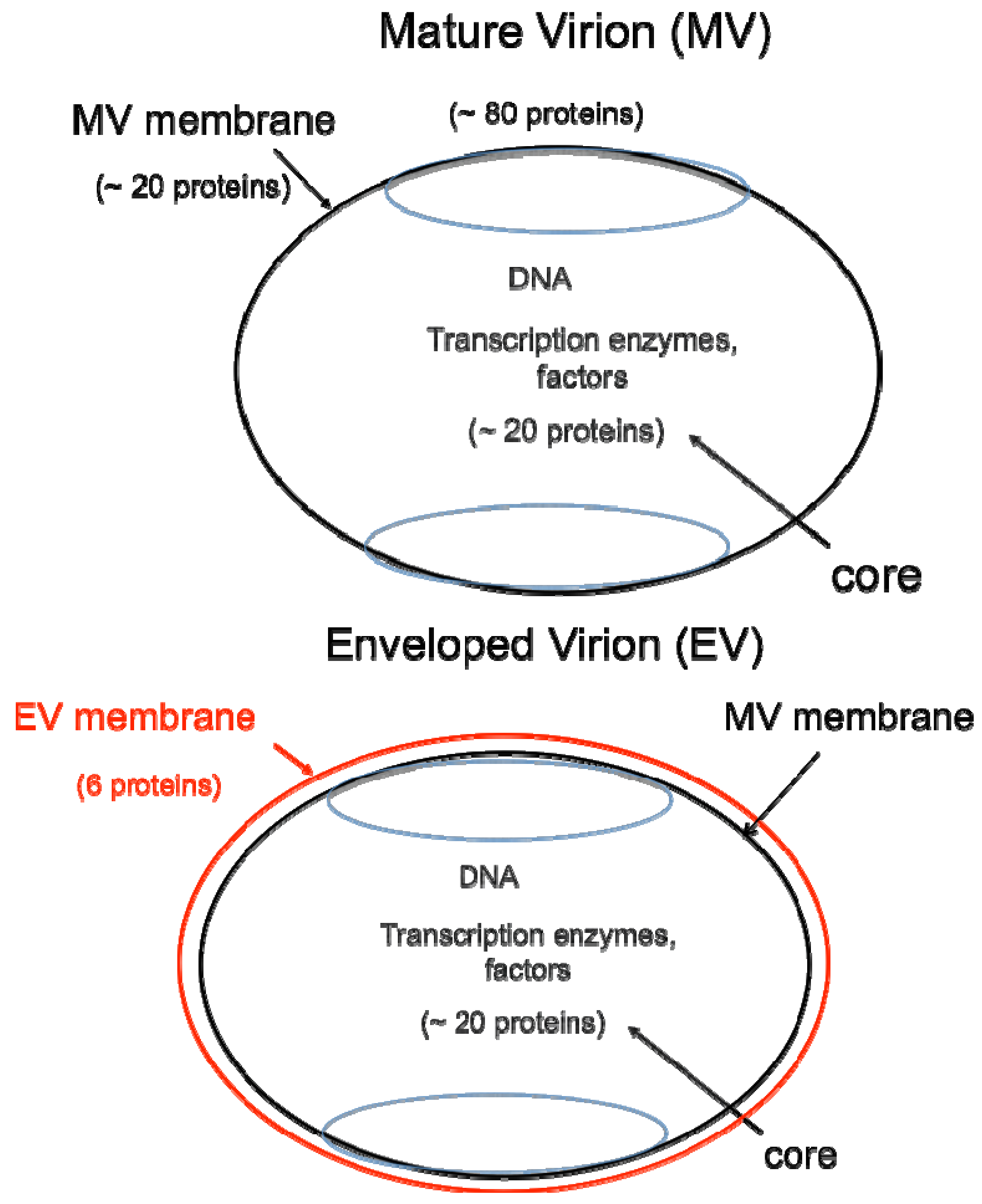
2. Poxvirus Replication Cycle

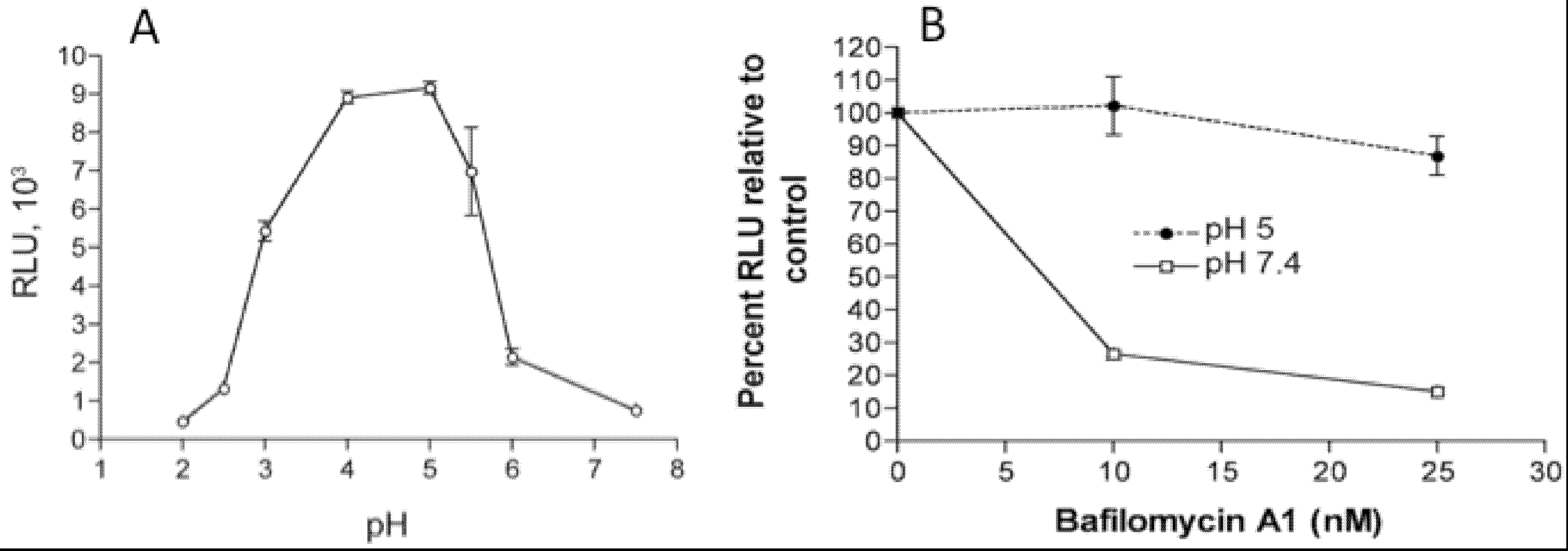
3. Entry Pathways


4. Attachment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | kDa | TM a | Expr b | Cons c | Properties |
|---|---|---|---|---|---|
| Attachment | |||||
| A26 | 58 | - | L | - | Binds laminin; assoc. with A27 |
| A27 | 13 | - | I | - | Binds heparan; assoc with A17; N d |
| D8 | 35 | N | I | - | Binds chondroitin; N |
| H3 | 38 | C | I | P | Binds heparan; N |
| Entry | |||||
| A16 | 43 | C | I | P | EFC e; paralog G9, J5; binds G9; C-C f |
| A21 | 14 | N | L | P | EFC; C-C |
| A28 | 16 | N | L | P | EFC; N; binds H2; C-C |
| F9 | 24 | C | L | P | EFC associated; C-C |
| G3 | 13 | N | L | P | EFC; binds L5 |
| G9 | 39 | C | L | P | EFC; paralog A16, J5; binds A16; C-C |
| H2 | 22 | N | L | P | EFC; binds A28; C-C |
| I2 | 8 | C | L | C | EFC? |
| J5 | 15 | C | L | P | EFC; paralog A16, G9; C-C |
| L1 | 27 | C | L | P | EFC associated; N; C-C; Myr g |
| L5 | 15 | C | L | P | EFC; binds G3; C-C |
| O3 | 4 | N | I | C | EFC |
5. Identification of Viral Proteins that Mediate Core Entry
6. Organization of the EFC and Structure of Subunit Proteins
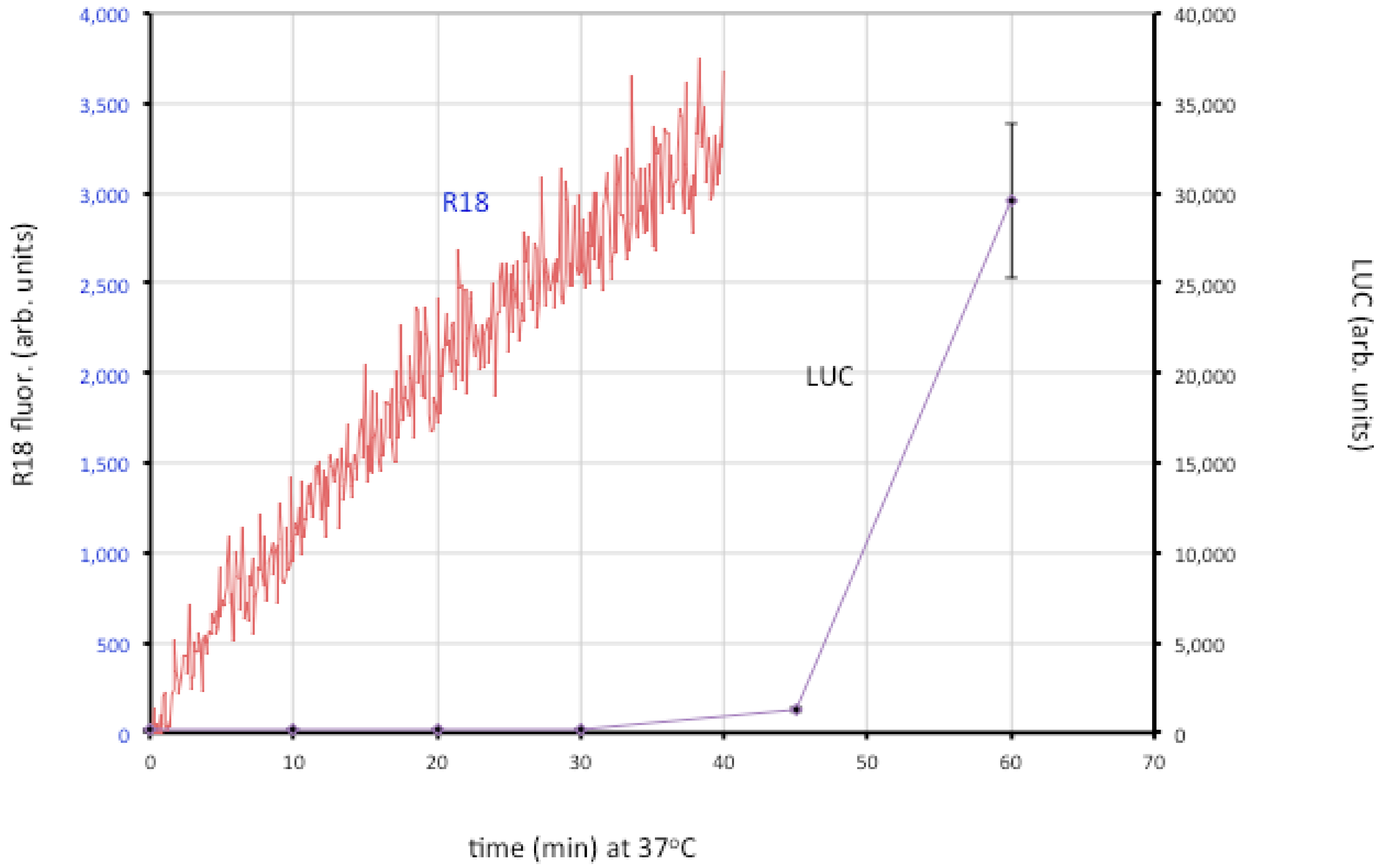
7. Membrane Fusion

8. Cell-Cell Fusion
9. Inhibition of Superinfection
10. Final Thoughts
Addendum
Acknowledgments
Conflict of Interest
References
- Moss, B. Poxviridae: The Viruses and Their Replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2905–2946. [Google Scholar]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar]
- Moss, B. Poxvirus entry and membrane fusion. Virology 2006, 344, 48–54. [Google Scholar]
- Moss, B.; de Silva, F. Poxvirus DNA Replication and Human Disease. In DNA Replication & Human Disease; DePamphilis, M.L., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; pp. 707–727. [Google Scholar]
- Yang, Z.; Reynolds, S.E.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 2011, 85, 9899–9908. [Google Scholar]
- Condit, R.C.; Moussatche, N.; Traktman, P. In a nutshell: Structure and assembly of the vaccinia virion. Adv. Virus Res. 2006, 66, 31–124. [Google Scholar]
- Hollinshead, M.; Vanderplasschen, A.; Smith, G.L.; Vaux, D.J. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 1999, 73, 1503–1517. [Google Scholar]
- Heuser, J. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “Honeycomb” Surface coat. J. Cell Biol. 2005, 169, 269–283. [Google Scholar]
- Cyrklaff, M.; Risco, C.; Fernandez, J.J.; Jimenez, M.V.; Esteban, M.; Baumeister, W.; Carrascosa, J.L. Cryo-electron tomography of vaccinia virus. Proc. Natl. Acad. Sci. USA 2005, 102, 2772–2777. [Google Scholar]
- Hiller, G.; Weber, K. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 1985, 55, 651–659. [Google Scholar]
- Schmelz, M.; Sodeik, B.; Ericsson, M.; Wolffe, E.J.; Shida, H.; Hiller, G.; Griffiths, G. Assembly of vaccinia virus: The second wrapping cisterna is derived from the trans Golgi network. J. Virol. 1994, 68, 130–147. [Google Scholar]
- Tooze, J.; Hollinshead, M.; Reis, B.; Radsak, K.; Kern, H. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 1993, 60, 163–178. [Google Scholar]
- Ward, B.M.; Moss, B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 2001, 75, 11651–11663. [Google Scholar]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar]
- Blasco, R.; Moss, B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 1992, 66, 4170–4179. [Google Scholar]
- Roper, R.; Wolffe, E.J.; Weisberg, A.; Moss, B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to- cell spread of vaccinia virus. J. Virol. 1998, 72, 4192–4204. [Google Scholar]
- Sanderson, C.M.; Frischknecht, F.; Way, M.; Hollinshead, M.; Smith, G.L. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 1998, 79, 1415–1425. [Google Scholar]
- Wolffe, E.J.; Katz, E.; Weisberg, A.; Moss, B. The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. J. Virol. 1997, 71, 3904–3915. [Google Scholar]
- Wolffe, E.J.; Weisberg, A.S.; Moss, B. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 1998, 244, 20–26. [Google Scholar]
- Payne, L.G. Significance of extracellular virus in the in vitro and in vivo dissemination of vaccinia virus. J. Gen. Virol. 1980, 50, 89–100. [Google Scholar]
- Blasco, R.; Sisler, J.R.; Moss, B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: Effect of a point mutation in the lectin homology domain of the A34R gene. J. Virol. 1993, 67, 3319–3325. [Google Scholar]
- Katz, E.; Ward, B.M.; Weisberg, A.S.; Moss, B. Mutations in the vaccinia virus A33R and B5R envelope proteins that enhance release of extracellular virions and eliminate formation of actin-dontaining microvilli without preventing tyrosine phosphorylation of the A36R protein. J. Virol. 2003, 77, 12266–12275. [Google Scholar]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar]
- Armstrong, J.A.; Metz, D.H.; Young, M.R. The mode of entry of vaccinia virus into L cells. J. Gen. Virol. 1973, 21, 533–537. [Google Scholar]
- Carter, G.C.; Law, M.; Hollinshead, M.; Smith, G.L. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 2005, 86, 1279–1290. [Google Scholar]
- Townsley, A.C.; Weisberg, A.S.; Wagenaar, T.R.; Moss, B. Vaccinia virus entry into cells via a low pH-dependent-endosomal pathway. J. Virol. 2006, 80, 8899–8908. [Google Scholar]
- Law, M.; Carter, G.C.; Roberts, K.L.; Hollinshead, M.; Smith, G.L. Ligand-induced and non-fusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 2006, 103, 5989–5994. [Google Scholar]
- Dales, S.; Siminovitch, L. The development of vaccinia virus in earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 1961, 10, 475–503. [Google Scholar]
- Dales, S.; Kajioka, R. The cycle of multiplication of vaccinia virus in earle’s strain L cells. I. Uptake and penetration. Virology 1964, 24, 278–294. [Google Scholar] [CrossRef]
- Townsley, A.C.; Moss, B. Two distinct low-pH steps promote entry of vaccinia virus. J. Virol. 2007, 81, 8613–8620. [Google Scholar]
- Bengali, Z.; Townsley, A.C.; Moss, B. Vaccinia virus strain differences in cell attachment and entry. Virology 2009, 389, 132–140. [Google Scholar]
- Villa, N.Y.; Bartee, E.; Mohamed, M.R.; Rahman, M.M.; Barrett, J.W.; McFadden, G. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 2010, 401, 266–279. [Google Scholar]
- Chang, S.J.; Chang, Y.X.; Izmailyan, R.; Tang, Y.L.; Chang, W. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into hela, CHO-K1, and L cells. J. Virol. 2010, 84, 8422–8432. [Google Scholar]
- Whitbeck, J.C.; Foo, C.-H.; Ponce de Leon, M.; Eisenberg, R.J.; Cohen, G.H. Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 2009, 385, 383–391. [Google Scholar]
- Li, Y.; Yuan, S.; Moyer, R.W. The non-permissive infection of insect (gypsy moth) LD-652 cells by vaccinia virus. Virology 1998, 248, 74–82. [Google Scholar]
- Moser, T.S.; Jones, R.G.; Thompson, C.B.; Coyne, C.B.; Cherry, S. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog. 2010, 6, e1000954. [Google Scholar]
- Bengali, Z.; Satheshkumar, P.S.; Yang, Z.; Weisberg, A.S.; Paran, N.; Moss, B. Drosophila S2 cells are non-permissive for vaccinia virus DNA replication following entry via low pH-dependent endocytosis and early transcription. PLoS One 2011, 6, e17248. [Google Scholar]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar]
- Huang, C.Y.; Lu, T.Y.; Bair, C.H.; Chang, Y.S.; Jwo, J.K.; Chang, W. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into hela cells through fluid phase endocytosis. J. Virol. 2008, 82, 7988–7999. [Google Scholar]
- Mercer, J.; Knebel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. USA 2010, 107, 9346–9351. [Google Scholar]
- Marsh, Y.V.; Eppstein, D.A. Vaccinia virus and the EGF receptor: A portal of entry for infectivity? J. Cell. Biochem. 1987, 34, 239–245. [Google Scholar] [CrossRef]
- Eppstein, D.A.; Marsh, Y.V.; Schreiber, A.B.; Newman, S.R.; Todaro, G.J.; Nestor, J.J. Epidermal growth factor receptor occupancy inhbits vaccinia virus infection. Nature (London) 1985, 318, 550–552. [Google Scholar] [CrossRef]
- Lalani, A.S.; Masters, J.; Zeng, W.; Barrett, J.; Pannu, R.; Everett, H.; Arendt, C.W.; McFadden, G. Use of chemokine receptors by poxviruses. Science 1999, 286, 1968–1971. [Google Scholar]
- Schroeder, N.; Chung, C.S.; Chen, C.H.; Liao, C.L.; Chang, W. The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J. Virol. 2012. [Google Scholar] [CrossRef]
- Sandgren, K.J.; Wilkinson, J.; Miranda-Saksena, M.; McInerney, G.M.; Byth-Wilson, K.; Robinson, P.J.; Cunningham, A.L. A differential role for macropinocytosis in mediating entry of the two forms of vaccinia virus into dendritic cells. PLoS Pathog. 2010, 6, e1000866. [Google Scholar]
- Schmidt, F.I.; Bleck, C.K.; Helenius, A.; Mercer, J. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 2011, 30, 3647–3661. [Google Scholar]
- Ichihashi, Y.; Oie, M. The activation of vaccinia virus infectivity by the transfer of phosphatidylserine from the plasma membrane. Virology 1983, 130, 306–317. [Google Scholar]
- Oie, M. Reversible inactivation and reactivation of vaccinia virus by manipulation of viral lipid composition. Virology 1985, 142, 299–306. [Google Scholar]
- Laliberte, J.P.; Moss, B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. USA 2009, 106, 17517–17521. [Google Scholar]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S. The soluble serum protein gGas6 bridges virion envelope phosphatidylserine to the tam receptor tyrosine kinase axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar]
- Vanderplasschen, A.; Smith, G.L. A novel virus binding assay using confocal microscopy: Demonstration that intracellular and extracellular vaccinia virions bind to different cellular receptors. J. Virol. 1997, 71, 4032–4041. [Google Scholar]
- Chahroudi, A.; Chavan, R.; Koyzr, N.; Waller, E.K.; Silvestri, G.; Feinberg, M.B. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. 2005, 79, 10397–10407. [Google Scholar]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999, 73, 8750–8761. [Google Scholar]
- Chung, C.-S.; Hsiao, J.-C.; Chang, Y.-S.; Chang, W. A27L protein mediates vaccinia virus interaction with cell surface heparin sulfate. J. Virol. 1998, 72, 1577–1585. [Google Scholar]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Cell surface proteoglycans are necessary for A27L protein- mediated cell fusion: Identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 1998, 72, 8374–8379. [Google Scholar]
- Lin, C.L.; Chung, C.S.; Heine, H.G.; Chang, W. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 2000, 74, 3353–3365. [Google Scholar]
- Vazquez, M.I.; Esteban, M. Identification of functional domains in the 14-kilodalton envelope protein (A27L) of vaccinia virus. J. Virol. 1999, 73, 9098–9109. [Google Scholar]
- Chiu, W.L.; Lin, C.L.; Yang, M.H.; Tzou, D.L.M.; Chang, W. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 2007, 81, 2149–2157. [Google Scholar]
- Rodriguez, D.; Rodriguez, J.R.; Esteban, M. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 1993, 67, 3435–3440. [Google Scholar]
- Howard, A.R.; Senkevich, T.G.; Moss, B. Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 2008, 82, 12384–12391. [Google Scholar]
- Ching, Y.C.; Chung, C.S.; Huang, C.Y.; Hsia, Y.; Tang, Y.L.; Chang, W. Disulfide bond formation at the C termini of vaccinia virus A26 andA27 proteins does not require viral redox enzymes and suppresses glycosaminoglycan-mediated cell fusion. J. Virol. 2009, 83, 6464–6476. [Google Scholar]
- da Fonseca, F.G.; Wolffe, E.J.; Weisberg, A.; Moss, B. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J. Virol. 2000, 74, 7518–7528. [Google Scholar]
- Ward, B.M. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 2005, 79, 4755–4763. [Google Scholar]
- McKelvey, T.A.; Andrews, S.C.; Miller, S.E.; Ray, C.A.; Pickup, D.J. Identification of the orthopoxvirus p4c gene, which encodes a structural protein that directs intracellular mature virus particles into A-type inclusions. J. Virol. 2002, 76, 11216–11225. [Google Scholar]
- Patel, D.D.; Pickup, D.J.; Joklik, W.K. Isolation of cowpox virus A-type inclusions and characterization of their major protein component. Virology 1986, 149, 174–189. [Google Scholar]
- Amegadzie, B.Y.; Sisler, J.R.; Moss, B. Frame-shift mutations within the vaccinia virus A-type inclusion protein gene. Virology 1992, 186, 777–782. [Google Scholar]
- Foo, C.H.; Lou, H.; Whitbeck, J.C.; Ponce de Leon, M.; Atanasiu, D.; Eisenberg, R.J.; Cohen, G.H. Vaccinia virus L1 binds to cell surfaces and blocks virus entry independently of glycosaminoglycans. Virology 2009, 385, 368–382. [Google Scholar]
- Satheshkumar, P.S.; Moss, B. Sequence-divergent chordopoxvirus homologs of the O3 protein maintain functional interactions with components of the vaccinia virus entry-fusion complex. J. Virol. 2012, 86, 1696–1705. [Google Scholar]
- Senkevich, T.G.; Ward, B.M.; Moss, B. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 2004, 78, 2357–2366. [Google Scholar]
- Townsley, A.; Senkevich, T.G.; Moss, B. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 2005, 79, 9458–9469. [Google Scholar]
- Townsley, A.; Senkevich, T.G.; Moss, B. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is requried for cell entry and cell-cell fusion. J. Virol. 2005, 79, 10988–10998. [Google Scholar]
- Senkevich, T.G.; Ojeda, S.; Townsley, A.; Nelson, G.E.; Moss, B. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 2005, 102, 18572–18577. [Google Scholar]
- Ojeda, S.; Senkevich, T.G.; Moss, B. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 2006, 80, 51–61. [Google Scholar]
- Senkevich, T.G.; Ward, B.M.; Moss, B. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 2004, 78, 2348–2356. [Google Scholar]
- Turner, P.C.; Dilling, B.P.; Prins, C.; Cresawn, S.G.; Moyer, R.W.; Condit, R.C. Vaccinia virus temperature-sensitive mutants in the A28 gene produce non-infectious virions that bind to cells but are defective in entry. Virology 2007, 366, 62–72. [Google Scholar]
- Izmailyan, R.A.; Huang, C.Y.; Mohammad, S.; Isaacs, S.N.; Chang, W. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J. Virol. 2006, 80, 8402–8410. [Google Scholar]
- Ojeda, S.; Domi, A.; Moss, B. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 2006, 80, 9822–9830. [Google Scholar]
- Senkevich, T.G.; Moss, B. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 2005, 79, 4744–4754. [Google Scholar]
- Nelson, G.E.; Wagenaar, T.R.; Moss, B. A conserved sequence within the H2 subunit of the vaccinia virus entry/fusion complex is important for interaction with the A28 subunit and infectivity. J. Virol. 2008, 82, 6244–6250. [Google Scholar]
- Wolfe, C.L.; Ojeda, S.; Moss, B. Transcriptional repression and RNA silencing act synergistically to demonstrate the function of the eleventh component of the vaccinia virus entry-fusion complex. J. Virol. 2012, 86, 293–301. [Google Scholar]
- Satheshkumar, P.S.; Moss, B. Characterization of a newly identified 35 amino acid component of the vaccinia virus entry/fusion complex conserved in all chordopoxviruses. J. Virol. 2009, 83, 12822–12832. [Google Scholar]
- Brown, E.; Senkevich, T.G.; Moss, B. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein. J. Virol. 2006, 80, 9455–9464. [Google Scholar]
- Bisht, H.; Weisberg, A.S.; Moss, B. Vaccinia virus L1 protein is required for cell entry and membrane fusion. J. Virol. 2008, 82, 8687–8694. [Google Scholar]
- Nichols, R.J.; Stanitsa, E.; Unger, B.; Traktman, P. The vaccinia I2L gene encodes a membrane protein with an essential role in virion entry. J. Virol. 2008, 82, 10247–10261. [Google Scholar]
- Kochan, G.; Escors, D.; Gonzalez, J.M.; Casasnovas, J.M.; Esteban, M. Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell. Microbiol. 2008, 10, 1149–1164. [Google Scholar]
- Rodríguez, D.; Esteban, M.; Rodríguez, J.R. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol. 1995, 69, 4640–4648. [Google Scholar]
- Wolffe, E.J.; Moore, D.M.; Peters, P.J.; Moss, B. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 1996, 70, 2797–2808. [Google Scholar]
- Wagenaar, T.R.; Ojeda, S.; Moss, B. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 2008, 82, 5153–5160. [Google Scholar]
- Wolfe, C.L.; Moss, B. Interaction between the G3 and L5 proteins of the vaccinia virus entry-fusion complex. Virology 2011, 412, 278–283. [Google Scholar]
- Senkevich, T.G.; White, C.L.; Koonin, E.V.; Moss, B. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. USA 2002, 99, 6667–6672. [Google Scholar]
- Ryser, H.J.; Levy, E.M.; Mandel, R.; DiSciullo, G.J. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. USA 1994, 91, 4559–4563. [Google Scholar]
- Markovic, I.; Stantchev, T.S.; Fields, K.H.; Tiffany, L.J.; Tomic, M.; Weiss, C.D.; Broder, C.C.; Strebel, K.; Clouse, K.A. Thiol/disulfide exchange is a pre-requisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry. Blood 2004, 103, 1586–1594. [Google Scholar]
- Wallin, M.; Ekstrom, M.; Garoff, H. The fusion-controlling disulfide bond isomerase in retrovirus Env is triggered by protein destabilization. J. Virol. 2005, 79, 1678–1685. [Google Scholar]
- Jain, S.; McGinnes, L.W.; Morrison, T.G. Thiol/disulfide exchange is required for membrane fusion directed by the newcastle disease virus fusion protein. J. Virol. 2007, 81, 2328–2339. [Google Scholar]
- Nelson, G.E.; Sisler, J.R.; Chandran, D.; Moss, B. Vaccinia virus entry/fusion complex subunit A28 is a target of neutralizing and protective antibodies. Virology 2008, 380, 394–401. [Google Scholar]
- Shinoda, K.; Wyatt, L.S.; Moss, B. The neutralizing antibody response to the vaccinia virus A28 protein is specifically enhanced by its association with the H2 protein. Virology 2010, 405, 41–49. [Google Scholar]
- Wolffe, E.J.; Vijaya, S.; Moss, B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 1995, 211, 53–63. [Google Scholar]
- Ichihashi, Y.; Oie, M. Neutralizing epitopes on penetration protein of vaccinia virus. Virology 1996, 220, 491–494. [Google Scholar]
- Franke, C.A.; Wilson, E.M.; Hruby, M.D. Use of a cell-free system to identify the vaccinia virus L1R gene product as the major late myristylated virion protein M25. J. Virol. 1990, 64, 5988–5996. [Google Scholar]
- Ravanello, M.P.; Hruby, D.E. Characterization of the vaccinia virus L1R myristylprotein as a component of the intracellular virion envelope. J. Gen. Virol. 1994, 75, 1479–1483. [Google Scholar]
- Aldaz-Carroll, L.; Whitbeck, J.C.; Ponce de Leon, M.; Lou, H.; Pannell, L.K.; Lebowitz, J.; Fogg, C.; White, C.; Moss, B.; Cohen, G.H.; et al. Physical and immunological characterization of a recombinant secreted form of the membrane protein encoded by the vaccinia virus L1R gene. Virology 2005, 341, 59–71. [Google Scholar] [CrossRef]
- Ravanello, M.P.; Hruby, D.E. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virus assembly. J. Virol. 1994, 68, 6401–6410. [Google Scholar]
- Bisht, H.; Brown, E.; Moss, B. Kinetics and intracellular location of intramolecular disulfide bond formation mediated by the cytoplasmic redox system encoded by vaccinia virus. Virology 2010, 398, 187–193. [Google Scholar]
- Foo, C.H.; Whitbeck, J.C.; Ponce de Leon, M.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. The myristate moiety and amino-terminus of the vaccinia virus L1 constitute a bipartite functional region needed for entry. J. Virol. 2012. [Google Scholar] [CrossRef]
- Su, H.P.; Garman, S.C.; Allison, T.J.; Fogg, C.; Moss, B.; Garboczi, D.N. The 1.51-Å structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 4240–4245. [Google Scholar]
- Su, H.P.; Golden, J.W.; Gittis, A.G.; Hooper, J.W.; Garboczi, D.N. Structural basis for the binding of the neutralizing antibody, 7D11, to the poxvirus L1 protein. Virology 2007, 368, 331–341. [Google Scholar]
- Doms, R.W.; Blumenthal, R.; Moss, B. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 1990, 64, 4884–4892. [Google Scholar]
- Laliberte, J.P.; Weisberg, A.S.; Moss, B. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog. 2011, 7, e1002446. [Google Scholar]
- Locker, J.K.; Kuehn, A.; Schleich, S.; Rutter, G.; Hohenberg, H.; Wepf, R.; Griffiths, G. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell 2000, 11, 2497–2511. [Google Scholar]
- Zheng, Q.A.; Chang, D.C. Reorganization of cytoplasmic structures during cell fusion. J. Cell Sci. 1991, 100, 431–442. [Google Scholar]
- Eitzen, G. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 2003, 1641, 175–181. [Google Scholar]
- Massarwa, R.; Carmon, S.; Shilo, B.Z.; Schejter, E.D. WIP/WASp-based actin-polymerization machinery is essential for myoblast fusion in Drosophila. Dev. Cell 2007, 12, 557–569. [Google Scholar]
- Kallewaard, N.L.; Bowen, A.L.; Crowe, J.E., Jr. Cooperativity of actin and microtubule elements during replication of respiratory syncytial virus. Virology 2005, 331, 73–81. [Google Scholar]
- Gower, T.L.; Pastey, M.K.; Peeples, M.E.; Collins, P.L.; McCurdy, L.H.; Hart, T.K.; Guth, A.; Johnson, T.R.; Graham, B.S. RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J. Virol. 2005, 79, 5326–5336. [Google Scholar]
- Pontow, S.E.; Heyden, N.V.; Wei, S.; Ratner, L. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J. Virol. 2004, 78, 7138–7147. [Google Scholar]
- Schowalter, R.M.; Wurth, M.A.; Aguilar, H.C.; Lee, B.; Moncman, C.L.; McCann, R.O.; Dutch, R.E. Rho GTPase activity modulates paramyxovirus fusion protein-mediated cell-cell fusion. Virology 2006, 350, 323–334. [Google Scholar]
- Stantchev, T.S.; Markovic, I.; Telford, W.G.; Clouse, K.A.; Broder, C.C. The tyrosine kinase inhibitor genistein blocks HIV-1 infection in primary human macrophages. Virus Res. 2007, 123, 178–189. [Google Scholar]
- Harmon, B.; Ratner, L. Induction of the Gαq signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J. Virol. 2008, 82, 9191–9205. [Google Scholar]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar]
- Ichihashi, Y.; Dales, S. Biogenesis of poxviruses: Interrelationship between hemagglutinin production and polykaryocytosis. Virology 1971, 46, 533–543. [Google Scholar]
- Turner, P.C.; Moyer, R.W. An orthopoxvirus serpin-like gene controls the ability of infected cells to fuse. J. Virol. 1992, 66, 2076–2085. [Google Scholar]
- Law, K.M.; Smith, G.L. A vaccinia serine protease inhibitor which prevents virus-induced cell fusion. J. Gen. Virol. 1992, 73, 549–557. [Google Scholar]
- Zhou, J.; Sun, X.Y.; Fernando, G.J.P.; Frazer, I.H. The vaccinia virus K2L gene encodes a serine protease inhibitor which inhibits cell-cell fusion. Virology 1992, 189, 678–686. [Google Scholar]
- Wagenaar, T.R.; Moss, B. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 2007, 81, 6286–6293. [Google Scholar]
- Turner, P.C.; Moyer, R.W. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology 2006, 347, 88–99. [Google Scholar]
- Wagenaar, T.R.; Moss, B. Expression of the A56 and K2 proteins is sufficient to inhibit vaccinia virus entry and cell fusion. J. Virol. 2009, 83, 1546–1554. [Google Scholar]
- Gong, S.C.; Lai, C.F.; Esteban, M. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 1990, 178, 81–91. [Google Scholar]
- Blasco, R.; Moss, B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000 dalton outer envelope protein. J. Virol. 1991, 65, 5910–5920. [Google Scholar]
- Vanderplasschen, A.; Hollinshead, M.; Smith, G.L. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 1998, 79, 877–887. [Google Scholar]
- Chang, S.J.; Shih, A.C.; Tang, Y.L.; Chang, W. Vaccinia mature virus fusion regulator A26 protein binds to A16 and G9 proteins of the viral entry fusion complex and dissociates from mature virions at low pH. J. Virol. 2012, 86, 3809–3818. [Google Scholar]
- Joklik, W.K. The intracellular uncoating of poxvirus DNA. I. The fate of radioactively-labeled rabbitpox virus. J. Mol. Biol. 1964, 8, 263–276. [Google Scholar] [CrossRef]
- Moss, B.; Rosenblum, E.N.; Grimley, P.M. Assembly of virus particles during mixed infection with wild-type vaccinia and a refampicin-resistant mutant. Virology 1971, 45, 135–148. [Google Scholar]
- Christen, L.; Seto, J.; Niles, E.G. Superinfection exclusion of vaccinia virus in virus-infected cell cultures. Virology 1990, 174, 35–42. [Google Scholar]
- Turner, P.C.; Moyer, R.W. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology 2008, 380, 226–233. [Google Scholar]
- Doceul, V.; Hollinshead, M.; van der Linden, L.; Smith, G.L. Repulsion of superinfecting virions: A mechanism for rapid virus spread. Science 2010, 327, 873–876. [Google Scholar]
- Schmidt, F.I.; Bleck, C.K.; Mercer, J. Poxvirus host cell entry. Curr. Opin. Virol. 2012, 2, 20–27. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moss, B. Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses 2012, 4, 688-707. https://doi.org/10.3390/v4050688
Moss B. Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses. 2012; 4(5):688-707. https://doi.org/10.3390/v4050688
Chicago/Turabian StyleMoss, Bernard. 2012. "Poxvirus Cell Entry: How Many Proteins Does it Take?" Viruses 4, no. 5: 688-707. https://doi.org/10.3390/v4050688
APA StyleMoss, B. (2012). Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses, 4(5), 688-707. https://doi.org/10.3390/v4050688