Genomic Sequences of Two Novel Levivirus Single-Stranded RNA Coliphages (Family Leviviridae): Evidence for Recombination in Environmental Strains

Abstract
:1. Introduction
2. Results
2.1. Sequence Analyses and Open Reading Frames

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genogroup | Source | Origin | Accession number |
|---|---|---|---|---|
| MS2 | I | sewage | Berkeley, CA | NC_001417 |
| M12 | I | sewage | Germany | AF195778 |
| DL1 | I | river water | Tijuana River, CA | EF107159 |
| DL2 | I | bay water | Delaware Bay, DE | N/A |
| DL13 | I | oyster | Whiskey Creek, NC | N/A |
| DL16 | I | bay water | Great Bay, NH | EF108464 |
| J20 | I | chicken litter | South Carolina | EF204939 |
| ST4 | I | unknown | unknown | EF204940 |
| R17 | I | sewage | Philadelphia, PA | EF108465 |
| fr | I | dung hill | Heidelberg, Germany | X15031 |
| DL52 | I-JS | bay water | Rachel Carson Reserve, NC | JQ966307 |
| DL54 | I-JS | bay water | Narragansett Bay, RI | JQ966308 |
| GA | II | sewage | Ookayama, Japan | NC_001426 |
| KU1 | II | sewage | Kuwait | AF227250 |
| DL10 | II | mussel | Tijuana River, CA | FJ483837 |
| DL20 | II | clam | Narragansett Bay, RI | FJ483839 |
| T72 | II | bird | Talbert Marsh sandflats, CA | FJ483838 |
| Open Reading Frame Locations (amino acids) | ||||||
|---|---|---|---|---|---|---|
| Strain | Group | Full length | ORF1 | ORF2 | ORF3 | ORF4 |
| MS2 a | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| M12 a,b | I | 3340b | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | ND |
| DL1 | I | 3570 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL2 | I | 3491c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL13 | I | 3491c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL16 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| J20 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| ST4 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| R17 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| fr a | I | 3575 | 129-1310(393) | 1336-1728(130) | 1691-1906(71) | 1762-3399(545) |
| DL52 | JS | 3525 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398 d (545) |
| DL54 | JS | 3398 c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398 d (545) |
| (i) Genogroup I and JS strains | ||
|---|---|---|
| Strain | DL52 | DL54 |
| DL52 | 100 | |
| DL54 | 96.73 | 100 |
| DL1 | 81.48 | 81.87 |
| DL16 | 85.41 | 84.72 |
| ST4 | 80.30 | 80.11 |
| R17 | 80.55 | 80.53 |
| J20 | 82.00 | 82.01 |
| MS2 | 80.12 | 80.01 |
| fr | 69.18 | 69.06 |
| (ii) Genogroup II and JS strains | ||
| Strain | DL52 | DL54 |
| DL52 | 100 | |
| DL54 | 96.73 | 100 |
| T72 | 53.96 | 53.53 |
| DL10 | 54.07 | 53.89 |
| DL20 | 52.87 | 52.65 |
| GA | 52.44 | 52.29 |
| KU1 | 52.94 | 52.66 |

2.2. Amino Acid Analysis
| Maturation Protein |
|---|
 |
| Capsid Protein |
 |
| Lysis Protein |
 |
| Replicase Protein |
|---|
 |
2.3. Pfam and Protein Sequence Motifs
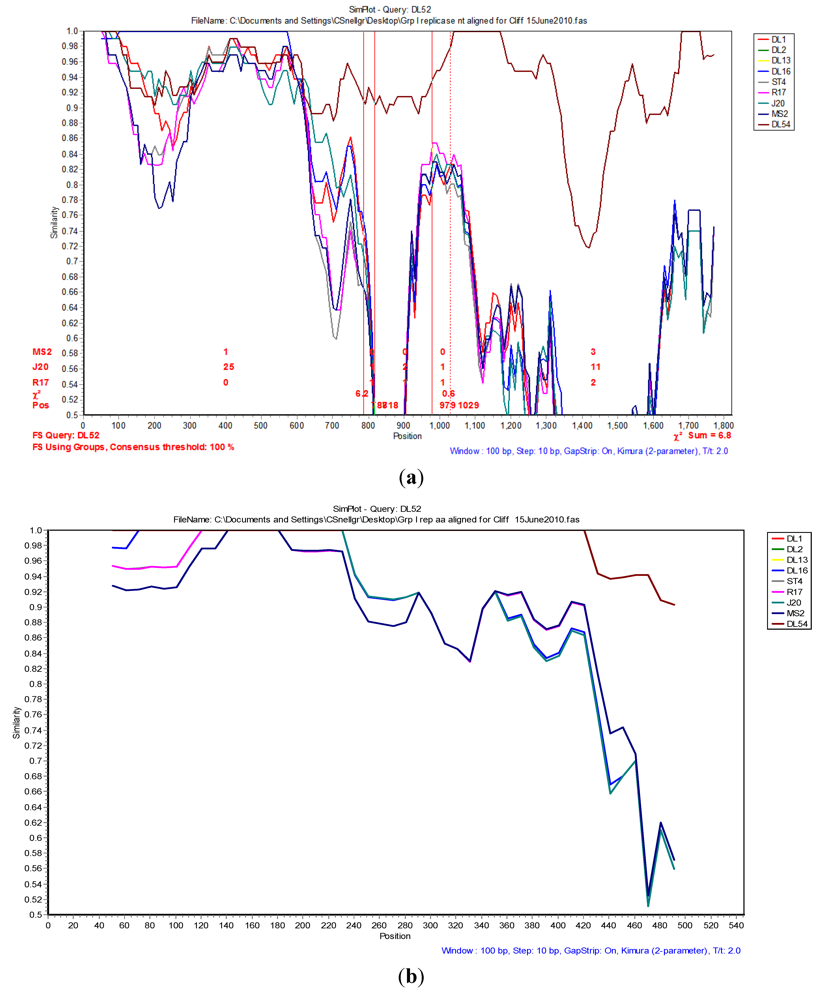
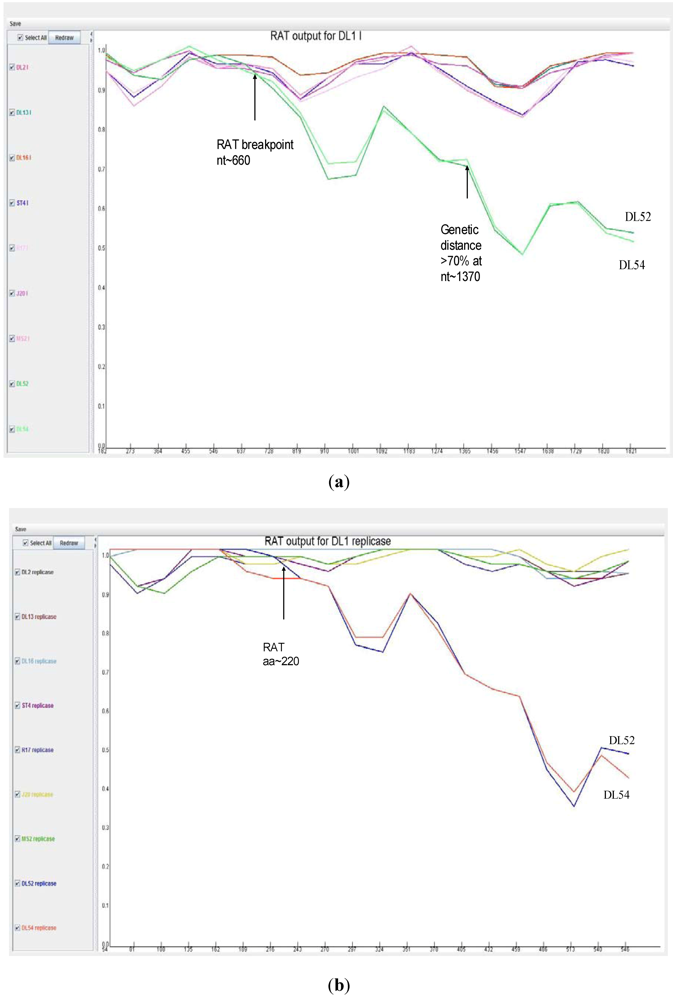
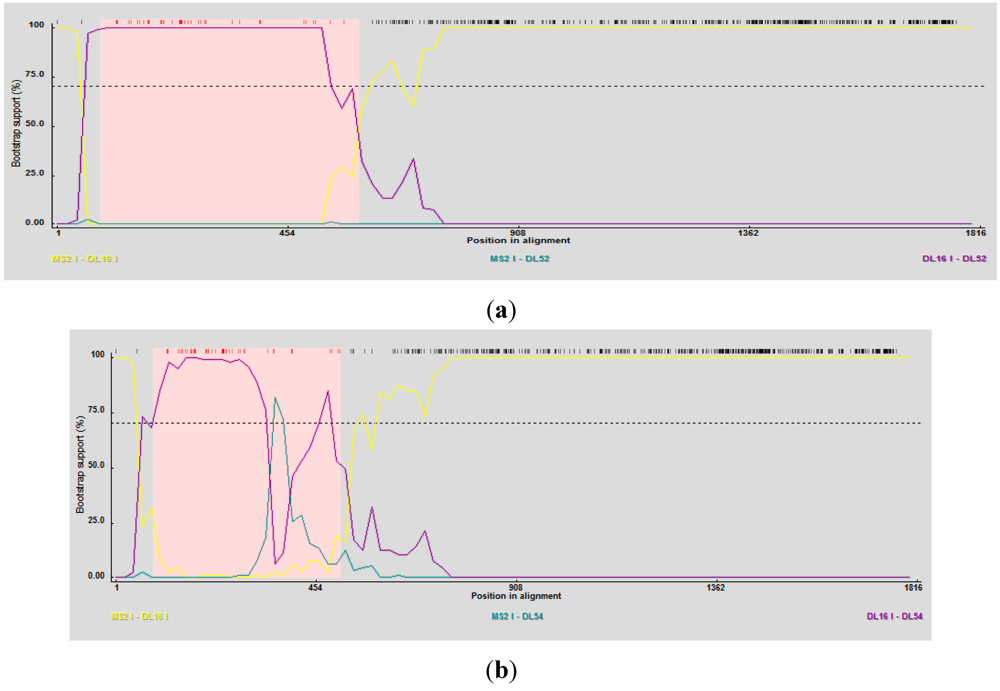
2.4. Phylogenetic and Recombination Analyses



| Confirmation Table of Recombination Events | ||
|---|---|---|
| Methods | Events | Average p-value |
| RDP | 2 | 2.199 × 10−15 |
| GENECONV | 1 | 3.031 × 10−27 |
| Bootscan | 2 | 7.867 × 10−19 |
| MaxChi | 2 | 1.445 × 10−10 |
| Chimaera | 2 | 3.536 × 10−11 |
| SiScan | 1 | 1.168 × 10−13 |
| 3Seq | 1 | 4.486 × 10−8 |


3. Discussion
4. Experimental Section
4.1. FRNA Coliphage Strains and RNA Extraction
4.2. Sequencing and Analysis
4.3. Amino Acid Analysis
4.4. Phylogenetic, Statistical and Recombination Analyses
4.5. Nucleotide Sequence Accession Numbers
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Fiers, W.; Contreras, R.; Duerinck, F.; Haegeman, G.; Iserentant, D.; Merregaert, J.; Min Jou, W.; Molemans, F.; Raeymaekers, A.; van den Berghe, A.; et al. Complete nucleotide sequence of bacteriophage MS2 RNA: Primary and secondary structure of the replicase gene. Nature 1976, 260, 500–507. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchinson, C.A. III; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage Ф-X174 DNA. Nature 1977, 265, 687–695. [Google Scholar]
- Friedman, S.D.; Genthner, F.J.; Gentry, J.; Sobsey, M.D; Vinjé, J. Gene mapping and phylogenetic analysis of the complete genome from 30 single-stranded RNA male-specific coliphages (family Leviviridae). J. Virol. 2009a, 83, 11233–11243. [Google Scholar]
- Vinjé, J.; Oudejans, S.J.G.; Stewart, J.R.; Sobsey, M.D.; Long, S.C. Molecular detection and genotyping of male-specific coliphages by reverse transcription-PCR and reverse line blot hybridization. Appl. Environ. Microbiol. 2004, 70, 5996–6004. [Google Scholar]
- Chetverin, A.B. The puzzle of RNA recombination. FEBS Lett. 1999, 460, 1–5. [Google Scholar] [CrossRef]
- Lutay, A.V.; Zenkova, M.A.; Vlassov, V.V. Nonenzymatic recombination of RNA: Possible mechanisms for the formation of novel sequences. Chem. Biodiv. 2007, 4, 762–767. [Google Scholar]
- Chetverin, A.B.; Kopein, D.S.; Chetverina, H.V.; Demidenko, A.A.; Ugarov, V.I. Viral RNA-directed RNA polymerases use diverse mechanisms to promote recombination between RNA molecules. J. Biol. Chem. 2005, 280, 8748–8755. [Google Scholar]
- Horiuchi, K. Genetic Studies of RNA Phages. In RNA Phages; Zinder, N.D., Ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1975; pp. 29–50. [Google Scholar]
- Munishkin, A.V.; Voronin, L.A.; Chetverin, A.B. An in vivo recombinant RNA capable of autocatalytic synthesis by Qβ replicase. Nature 1988, 333, 473–475. [Google Scholar] [CrossRef]
- Sobsey, M.D.; Love, D.C.; Lovelace, G.L. F+RNA coliphages as source tracking viral indicators of fecal pollution. A final report submitted to the NOAA/UNH Cooperative Institute for Coastal and Estuarine Environmental Technology (CICEET). 2006. [Google Scholar]
- Love, D.C.; Vinjé, J.; Khalil, S.M.; Murphy, J.; Lovelace, G.L.; Sobsey, M.D. Evaluation of RT-PCR and reverse line blot hybridization for detection and genotyping F+ RNA coliphages from estuarine waters and molluscan shellfish. J. Appl. Microbiol. 2008, 104, 1203–1212. [Google Scholar] [CrossRef]
- Lai, M.M.C. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar]
- Cristina, J.; Colina, R. Evidence of structural genomic region recombination in Hepatitis C virus. Virol. J. 2006, 3, 53–60. [Google Scholar] [CrossRef]
- Pantin-Jackwood, M.J.; Spackman, E.; Woolcock, P.R. Phylogenetic analysis of turkey astroviruses reveals evidence of recombination. Virus Genes 2006, 32, 187–192. [Google Scholar] [CrossRef]
- Holmes, E.C.; Worobey, M.; Rambaut, A. Phylogenetic evidence for recombination in Dengue virus. Mol. Biol. Evol. 1999, 16, 405–409. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Evidence for frequent recombination within species Human Enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J. Virol. 2004, 78, 855–867. [Google Scholar] [CrossRef]
- Banner, L.R.; Lai, M.M.C. Random nature of coronavirus RNA recombination in the absence of selection pressure. Virolgy 1991, 185, 441–445. [Google Scholar] [CrossRef]
- Oprisan, G.; Combiescu, M.; Guillot, S.; Caro, V.; Combiescu, A.; Delpeyroux, F.; Crainic, R. Natural genetic recombination between co-circulating heterotypic enteroviruses. J. Gen. Virol. 2002, 83, 2193–2200. [Google Scholar]
- Walter, J.E.; Briggs, J.; Guerrero, M.L.; Matson, D.O.; Pickering, L.K.; Ruiz-Palacios, G.; Berke, T.; Mitchell, D.K. Molecular characterization of a novel recombinant strain of human astrovirus associated with gastroenteritis in children. Arch. Virol. 2001, 146, 2357–2367. [Google Scholar] [CrossRef]
- Jiang, X.; Espul, N.; Zhong, W.M.; Cuello, H.; Matson, D.O. Characterization of a novel human calicivirus that may be a naturally occurring recombinant. Arch. Virol. 1999, 144, 2477–2387. [Google Scholar]
- Belliot, G.; Laveran, H.; Monroe, S.S. Detection and genetic differentiation of human astroviruses: Phylogenetic grouping varies by coding region. Arch. Virol. 1997, 142, 1323–1334. [Google Scholar] [CrossRef]
- Hirst, G.K. Genetic recombination with Newcastle disease virus, polioviruses, and influenza. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 303–309. [Google Scholar] [CrossRef]
- Ledinko, N. Genetic recombination with poliovirus type 1 studies of crosses between a normal horse serum-resistant mutant and several guanidine-resistant mutants of the same strain. Virology 1963, 20, 107–119. [Google Scholar] [CrossRef]
- Kew, O.; Morris-Glasgow, V.; Landaverde, M.; Burns, C.; Shaw, J.; Garib, Z.; André, J.; Blackman, E.; Freeman, C.J.; Jorba, J.; et al. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 2002, 296, 356–359. [Google Scholar] [CrossRef]
- Lee, W.H.; Sung, W.K. RB-finder: An improved distance-based sliding window method to detect recombination breakpoints. J. Comput. Biol. 2008, 15, 881–898. [Google Scholar] [CrossRef]
- Stewart, J.R.; Vinjé, J.; Oudejans, S.J.G.; Scott, G.I. Sequence variation among group III F-specific RNA coliphages from water samples and swine lagoons. Appl. Environ. Microbiol. 2006, 72, 1226–1230. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: New York, NY, USA, 1969; pp. 21–132. [Google Scholar]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Etherington, G.J.; Dicks, J.; Roberts, I.N. Recombination analysis tool (RAT): A program for the high-throughput detection of recombination. Bioinformatics 2005, 21, 278–281. [Google Scholar] [CrossRef]
- Martin, D.P.; Williamson, C.; Posada, D. RDP2: Recombination detection and analysis from sequence alignments. Bioinformatics 2005, 21, 260–262. [Google Scholar] [CrossRef]
- Maydt, J.; Lengauer, T. Recco: Recombination analysis using cost optimization. Bioinformatics 2006, 22, 1064–1071. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Friedman, S.D.; Snellgrove, W.C.; Genthner, F.J. Genomic Sequences of Two Novel Levivirus Single-Stranded RNA Coliphages (Family Leviviridae): Evidence for Recombination in Environmental Strains. Viruses 2012, 4, 1548-1568. https://doi.org/10.3390/v4091548
Friedman SD, Snellgrove WC, Genthner FJ. Genomic Sequences of Two Novel Levivirus Single-Stranded RNA Coliphages (Family Leviviridae): Evidence for Recombination in Environmental Strains. Viruses. 2012; 4(9):1548-1568. https://doi.org/10.3390/v4091548
Chicago/Turabian StyleFriedman, Stephanie D., Wyatt C. Snellgrove, and Fred J. Genthner. 2012. "Genomic Sequences of Two Novel Levivirus Single-Stranded RNA Coliphages (Family Leviviridae): Evidence for Recombination in Environmental Strains" Viruses 4, no. 9: 1548-1568. https://doi.org/10.3390/v4091548
APA StyleFriedman, S. D., Snellgrove, W. C., & Genthner, F. J. (2012). Genomic Sequences of Two Novel Levivirus Single-Stranded RNA Coliphages (Family Leviviridae): Evidence for Recombination in Environmental Strains. Viruses, 4(9), 1548-1568. https://doi.org/10.3390/v4091548