Foamy Virus Vectors for HIV Gene Therapy

Abstract
:1. HIV and HAART: Limitations of the Current Standard of Care
2. HIV Gene Therapy
4. Limitations of LV Vectors
5. FV Vectors


6. Vector Genotoxicity
6.2. Dysregulation of Neighboring Genes
6.3. Vector Design to Reduce Genotoxicity
7. FV Vector HSC Gene Therapy Models
8. FV Vector Anti-HIV Studies
8.1. In Vitro Studies

{kind=link}
{kind=link}
| Transgene | Description | Efficacy | Promoter | Assay | Publication |
|---|---|---|---|---|---|
| R2 | SIV rev + env shRNA | 68%–80% inhibition of viral replication | U6 | SIV challenge, CEMx174 cell line | Park et al. 2005 [77] |
| L2R | HIV LTR + rev miRNA cassette | >98% inhibition of viral replication | CMV | HIV challenge, U87.CD4.CXCR4 cell line | Park et al. 2009 [78] |
| TAR + L2R | Tat inducible HIV LTR + rev miRNA cassette + TAR | >98% inhibition of viral replication | Tat inducible LTR-Hsp fusion | ||
| TAR + R | Tat inducible rev miRNA cassette + TAR | >98% inhibition of viral replication | Tat-inducible LTR-Hsp fusion | ||
| TAR | TAR | >98% inhibition of viral replication | LTR | ||
| Sh1 | anti tat/rev shRNA | 4 log reduction of viral replication | U6 | HIV challenge, CD34-derived macrophages | Taylor et al. 2008 [79] |
| C46 | membrane associated fusion inhibitor | 4 log reduction of viral replication | MSCV | ||
| Sh1 + C46 + RevM10 | tat/rev shRNA + membrane-associated fusion inhibitor + dominant negative Rev | significantly increased relative to C46 alone | U6, MSCV, PGK | HIV challenge of protected and unprotected cells in CEMx174 cell line | |
| C46 | membrane associated fusion inhibitor | 5.2-fold increase in cell survival +3.1-fold decrease in HIV p24/cell | MSCV | ||
| 4 log reduction of viral replication | SFFV | SHIV challenge, CEM.NKR-CCR5 lymphocytes | Kiem et al. 2010 [27] | ||
| 15–20 fold reduction of viral replication | SFFV | SHIV or HIV single viral cycle challenge, MAGI-CCR5 cell line | |||
| SI + C46 | tat/rev shRNA + membrane associated fusion inhibitor | 5 fold reduction of viral replication | U6, SFFV | ||
| SII + SI + R5 + C46 | two tat/rev shRNAs + CCR5 shRNA + membrane associated fusion inhibitor | 23 fold reduction of viral replication | H1, SFFV | ||
| 4 log reduction of viral replication | SHIV challenge, CEM.NKR-CCR5 lymphocytes | ||||
| SI + C46 | tat/rev shRNA + C46 | 4 log reduction of viral replication | U6, SFFV | ||
| SII + SI + R5 | two tat/rev shRNAs + CCR5 shRNA | 180 fold reduction of viral replication | H1 |
8.2. In Vivo Selection of Human SCID Repopulating Cells
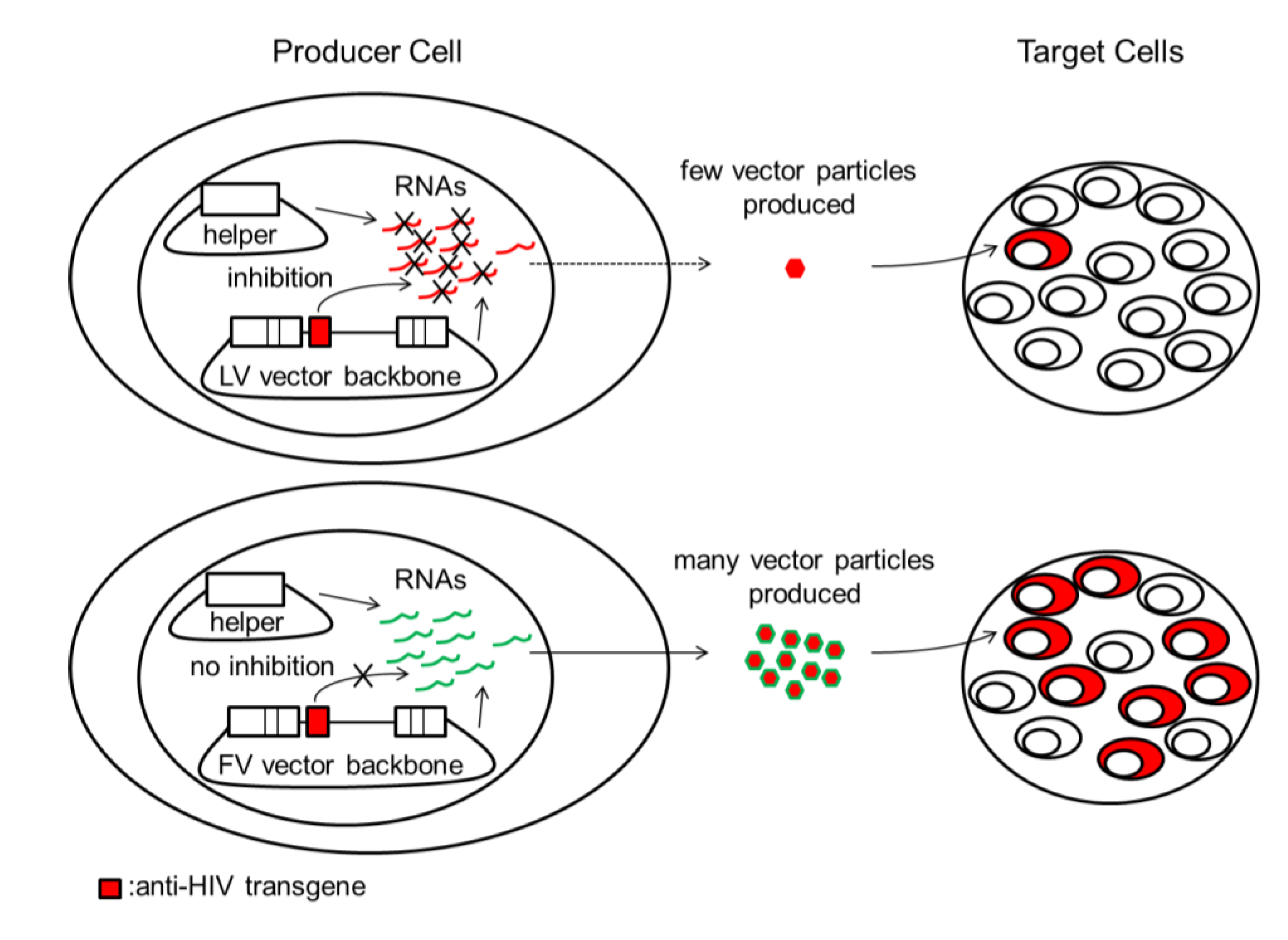
8.3. Anti-HIV shRNAs Inhibit LV but Not FV Vector Production
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- WHO, Global HIV/AIDS Response. Epidemic Update and Health Sector Progress towards Universal Access Progress Report 2011; WHO Press: Geneva, Switzerland, 2011.
- Ray, M.; Logan, R.; Sterne, J.A.; Hernandez-Diaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; van Sighem, A.; de Wolf, F.; Costagliola, D.; et al. The effect of combined antiretroviral therapy on the overall mortality of hiv-infected individuals. AIDS 2010, 24, 123–137. [Google Scholar] [CrossRef]
- Altmann, M.; An der Heiden, M.; Scheufele, R.; Hartmann, K.; Houareau, C.; Bartmeyer, B.; Hamouda, O.; German, H.I.V.S.C. The risk of aids-defining events is decreasing over time in the german HIV-1 seroconverter cohort. BMC Infect. Dis. 2012, 12, e94. [Google Scholar]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef]
- Valentin, A.; Morrow, M.; Poirier, R.H.; Aleman, K.; Little, R.; Yarchoan, R.; Pavlakis, G.N. Identification of a potential pharmacological sanctuary for HIV Type 1 in a fraction of CD4(+) primary cells. AIDS Res. Hum. Retroviruses 2010, 26, 79–88. [Google Scholar] [CrossRef]
- Delobel, P.; Sandres-Saune, K.; Cazabat, M.; L’Faqihi, F.E.; Aquilina, C.; Obadia, M.; Pasquier, C.; Marchou, B.; Massip, P.; Izopet, J. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. AIDS 2005, 19, 1739–1750. [Google Scholar] [CrossRef]
- Yukl, S.A.; Shergill, A.K.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Gunthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef]
- Lambotte, O.; Chaix, M.L.; Gubler, B.; Nasreddine, N.; Wallon, C.; Goujard, C.; Rouzioux, C.; Taoufik, Y.; Delfraissy, J.F. The lymphocyte HIV reservoir in patients on long-term HAART is a memory of virus evolution. AIDS 2004, 18, 1147–1158. [Google Scholar] [CrossRef]
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Richman, D.D. Update of the drug resistance mutations in HIV-1: March 2013. Top. Antivir. Med. 2013, 21, 6–14. [Google Scholar]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef]
- Maldarelli, F.; Palmer, S.; King, M.S.; Wiegand, A.; Polis, M.A.; Mican, J.; Kovacs, J.A.; Davey, R.T.; Rock-Kress, D.; Dewar, R.; et al. Art suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007, 3, e46. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef]
- Smith, R.L.; de Boer, R.; Brul, S.; Budovskaya, Y.; van Spek, H. Premature and accelerated aging: HIV or HAART? Front. Genet. 2012, 3, e328. [Google Scholar]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta 2006, 1757, 700–707. [Google Scholar]
- Fernandez-Fernandez, B.; Montoya-Ferrer, A.; Sanz, A.B.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Poveda, J.; Sainz-Prestel, V.; Ortiz-Martin, N.; Parra-Rodriguez, A.; Selgas, R.; et al. Tenofovir nephrotoxicity: 2011 update. AIDS Res. Treat. 2011, 2011, e354908. [Google Scholar]
- Gebo, K.A.; Fleishman, J.A.; Conviser, R.; Hellinger, J.; Hellinger, F.J.; Josephs, J.S.; Keiser, P.; Gaist, P.; Moore, R.D.; Network, H.I.V.R. Contemporary costs of HIV healthcare in the HAART era. AIDS 2010, 24, 2705–2715. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Zack, J.A.; Macpherson, J.L.; Symonds, G.P. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: Lessons learnt. Stem Cells Int. 2011, 2011, e393698. [Google Scholar]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef]
- Bokhoven, M.; Stephen, S.L.; Knight, S.; Gevers, E.F.; Robinson, I.C.; Takeuchi, Y.; Collins, M.K. Insertional gene activation by lentiviral and gammaretroviral vectors. J. Virol. 2009, 83, 283–294. [Google Scholar] [CrossRef]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-ccr5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez de Arellano, E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 15, 76–85. [Google Scholar] [CrossRef]
- Mautino, M.R.; Morgan, R.A. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mrna. Hum. Gene Ther. 2000, 11, 2025–2037. [Google Scholar] [CrossRef]
- Logan, A.C.; Haas, D.L.; Kafri, T.; Kohn, D.B. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J. Virol. 2004, 78, 8421–8436. [Google Scholar] [CrossRef]
- Morris, K.V.; Looney, D.J. Characterization of human immunodeficiency virus (HIV)-2 vector mobilization by HIV-1. Hum. Gene Ther. 2005, 16, 1463–1472. [Google Scholar] [CrossRef]
- Turner, A.M.; de La Cruz, J.; Morris, K.V. Mobilization-competent lentiviral vector-mediated sustained transcriptional modulation of HIV-1 expression. Mol. Ther. 2009, 17, 360–368. [Google Scholar] [CrossRef]
- Poeschla, E.M.; Wong-Staal, F.; Looney, D.J. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 1998, 4, 354–357. [Google Scholar] [CrossRef]
- Mitrophanous, K.; Yoon, S.; Rohll, J.; Patil, D.; Wilkes, F.; Kim, V.; Kingsman, S.; Kingsman, A.; Mazarakis, N. Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther. 1999, 6, 1808–1818. [Google Scholar] [CrossRef]
- Siapati, E.K.; Bigger, B.W.; Miskin, J.; Chipchase, D.; Parsley, K.L.; Mitrophanous, K.; Themis, M.; Thrasher, A.J.; Bonnet, D. Comparison of HIV- and EIAV-based vectors on their efficiency in transducing murine and human hematopoietic repopulating cells. Mol. Ther. 2005, 12, 537–546. [Google Scholar] [CrossRef]
- Price, M.A.; Case, S.S.; Carbonaro, D.A.; Yu, X.J.; Petersen, D.; Sabo, K.M.; Curran, M.A.; Engel, B.C.; Margarian, H.; Abkowitz, J.L.; et al. Expression from second-generation feline immunodeficiency virus vectors is impaired in human hematopoietic cells. Mol. Ther. 2002, 6, 645–652. [Google Scholar] [CrossRef]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog. 2012, 8, e1002790. [Google Scholar] [CrossRef]
- Betsem, E.; Rua, R.; Tortevoye, P.; Froment, A.; Gessain, A. Frequent and recent human acquisition of simian foamy viruses through apes’ bites in central Africa. PLoS Pathog. 2011, 7, e1002306. [Google Scholar] [CrossRef]
- Switzer, W.M.; Bhullar, V.; Shanmugam, V.; Cong, M.E.; Parekh, B.; Lerche, N.W.; Yee, J.L.; Ely, J.J.; Boneva, R.; Chapman, L.E.; et al. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 2004, 78, 2780–2789. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar]
- Ho, Y.P.; Schnabel, V.; Swiersy, A.; Stirnnagel, K.; Lindemann, D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol. Ther. 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
- Flugel, R.M. Spumaviruses: A group of complex retroviruses. J. Acquir. Immune Defic. Syndr. 1991, 4, 739–750. [Google Scholar]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Allen, J.; Peterson, L.; Ironside, C.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef]
- Tisdale, J.F.; Hanazono, Y.; Sellers, S.E.; Agricola, B.A.; Metzger, M.E.; Donahue, R.E.; Dunbar, C.E. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood 1998, 92, 1131–1141. [Google Scholar]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to evi1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. Bet proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Gijsbers, R.; Vets, S.; de Rijck, J.; Ocwieja, K.E.; Ronen, K.; Malani, N.; Bushman, F.D.; Debyser, Z. Role of the PWWP domain of lens epithelium-derived growth factor (LEDGF)/p75 cofactor in lentiviral integration targeting. J. Biol. Chem. 2011, 286, 41812–41825. [Google Scholar] [CrossRef]
- Vets, S.; de Rijck, J.; Brendel, C.; Grez, M.; Bushman, F.; Debyser, Z.; Gijsbers, R. Transient expression of an LEDGF/p75 chimera retargets lentivector integration and functionally rescues in a model for X-CGD. Mol. Ther. 2013, 2, e77. [Google Scholar]
- Rae, D.T.; Trobridge, G.D. Retroviral Genotoxicity. In Gene therapy—Tools and Potential Applications; Martin, F., Ed.; InTech: Rijeka, Croatia, 2013; pp. 399–427. [Google Scholar]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Invest. 2012, 122, 1667–1676. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Maroney, P.A.; Goodwin, R.G.; Rottman, F.M.; Crittenden, L.B.; Raines, M.A.; Kung, H.J. C-erbB activation in ALV-induced erythroblastosis: Novel RNA processing and promoter insertion result in expression of an amino-truncated EGF receptor. Cell 1985, 41, 719–726. [Google Scholar] [CrossRef]
- Li, C.L.; Xiong, D.; Stamatoyannopoulos, G.; Emery, D.W. Genomic and functional assays demonstrate reduced gammaretroviral vector genotoxicity associated with use of the CHS4 chromatin insulator. Mol. Ther. 2009, 17, 716–724. [Google Scholar] [CrossRef]
- Schambach, A.; Galla, M.; Maetzig, T.; Loew, R.; Baum, C. Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol. Ther. 2007, 15, 1167–1173. [Google Scholar]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Josephson, N.C.; Vassilopoulos, G.; Trobridge, G.D.; Priestley, G.V.; Wood, B.L.; Papayannopoulou, T.; Russell, D.W. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc. Natl. Acad. Sci. USA 2002, 99, 8295–8300. [Google Scholar] [CrossRef]
- Zucali, J.R.; Ciccarone, T.; Kelley, V.; Park, J.; Johnson, C.M.; Mergia, A. Transduction of umbilical cord blood CD34+ NOD/SCID-repopulating cells by simian foamy virus type 1 (SFV-1) vector. Virology 2002, 302, 229–235. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Kiem, H.P. Large animal models of hematopoietic stem cell gene therapy. Gene Ther. 2010, 17, 939–948. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Horn, P.A.; Beard, B.C.; Kiem, H.P. Large animal models for foamy virus vector gene therapy. Viruses 2012, 4, 3572–3588. [Google Scholar] [CrossRef]
- Suter, S.E.; Gouthro, T.A.; McSweeney, P.A.; Nash, R.A.; Haskins, M.E.; Felsburg, P.J.; Henthorn, P.S. Isolation and characterization of pediatric canine bone marrow CD34+ cells. Vet. Immunol. Immunopathol. 2004, 101, 31–47. [Google Scholar] [CrossRef]
- Creevy, K.E.; Bauer, T.R., Jr.; Tuschong, L.M.; Embree, L.J.; Silverstone, A.M.; Bacher, J.D.; Romines, C.; Garnier, J.; Thomas, M.L. Mixed chimeric hematopoietic stem cell transplant reverses the disease phenotype in canine leukocyte adhesion deficiency. Vet. Immunol. Immunopathol. 2003, 95, 113–121. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Gu, Y.C.; Tuschong, L.M.; Burkholder, T.; Bacher, J.D.; Starost, M.F.; Donahue, R.E.; Sokolic, R.A.; Hickstein, D.D. Nonmyeloablative hematopoietic stem cell transplantation corrects the disease phenotype in the canine model of leukocyte adhesion deficiency. Exp. Hematol. 2005, 33, 706–712. [Google Scholar] [CrossRef]
- Zaucha, J.A.; Yu, C.; Lothrop, C.D., Jr.; Nash, R.A.; Sale, G.; Georges, G.; Kiem, H.P.; Niemeyer, G.P.; Dufresne, M.; Cao, Q.; et al. Severe canine hereditary hemolytic anemia treated by nonmyeloablative marrow transplantation. Biol. Blood Marrow Transplant. 2001, 7, 14–24. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS One 2012, 7, e45173. [Google Scholar]
- Bauer, T.R., Jr.; Tuschong, L.M.; Calvo, K.R.; Shive, H.R.; Burkholder, T.H.; Karlsson, E.K.; West, R.R.; Russell, D.W.; Hickstein, D.D. Long-term follow-up of foamy viral vector-mediated gene therapy for canine leukocyte adhesion deficiency. Mol. Ther. 2013, 21, 964–972. [Google Scholar] [CrossRef]
- Park, J.; Nadeau, P.; Zucali, J.R.; Johnson, C.M.; Mergia, A. Inhibition of simian immunodeficiency virus by foamy virus vectors expressing sirnas. Virology 2005, 343, 275–282. [Google Scholar] [CrossRef]
- Park, J.; Nadeau, P.E.; Mergia, A. Activity of TAR in inducible inhibition of HIV replication by foamy virus vector expressing siRNAs under the control of HIV LTR. Virus Res. 2009, 140, 112–120. [Google Scholar] [CrossRef]
- Taylor, J.A.; Vojtech, L.; Bahner, I.; Kohn, D.B.; Laer, D.V.; Russell, D.W.; Richard, R.E. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Mol. Ther. 2008, 16, 46–51. [Google Scholar] [CrossRef]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef]
- Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Isolation of human O6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O6-benzylguanine. Cancer Res. 1998, 58, 1936–1945. [Google Scholar]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Invest. 2010, 120, 2345–2354. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olszko, M.E.; Trobridge, G.D. Foamy Virus Vectors for HIV Gene Therapy. Viruses 2013, 5, 2585-2600. https://doi.org/10.3390/v5102585
Olszko ME, Trobridge GD. Foamy Virus Vectors for HIV Gene Therapy. Viruses. 2013; 5(10):2585-2600. https://doi.org/10.3390/v5102585
Chicago/Turabian StyleOlszko, Miles E., and Grant D. Trobridge. 2013. "Foamy Virus Vectors for HIV Gene Therapy" Viruses 5, no. 10: 2585-2600. https://doi.org/10.3390/v5102585
APA StyleOlszko, M. E., & Trobridge, G. D. (2013). Foamy Virus Vectors for HIV Gene Therapy. Viruses, 5(10), 2585-2600. https://doi.org/10.3390/v5102585