Learning from the Messengers: Innate Sensing of Viruses and Cytokine Regulation of Immunity — Clues for Treatments and Vaccines

Abstract
:Abbreviations
| AIDS | Acquired immune deficiency syndrome |
| ADAR1 | Adenosine deaminase acting on RNA 1 |
| AdV | Adenovirus |
| AIM2 | Absent in melanoma 2 |
| AP-1 | Activator protein 1 |
| APOBEC3 | Apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3 |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment domain |
| ATF2 | Activating transcription factor 2 |
| AZT | Azidothymidine |
| BMDC | Bone marrow-derived DC |
| CARD | Caspase recruitment domain |
| CCL5 | CC chemokine ligand 5 (previously known as regulated upon activation, normal T cell expressed and secreted (RANTES)) |
| CCR5 | CC chemokine receptor 5 |
| CMV | Cytomegalovirus |
| CLR | C-type lectin receptor |
| CXCL10 | CXC chemokine ligand 10 |
| CYPA | Cyclophilin A |
| DAI | DNA-dependent activator of IFN-regulatory factors |
| DAMP | Danger-associated molecular pattern |
| DC | Dendritic cell |
| DC-SIGN | Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin |
| DDX41 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 41 |
| DHX9 | DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 9 |
| Ds | Double-stranded |
| EBV | Epstein Barr virus |
| E.Coli | Escherichia coli |
| EMCV | Encephalo myocarditis virus |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| Flu | Influenza virus |
| GAS | IFN-γ-activated site |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| HBV | Hepatitis B virus |
| HBsAg | Hepatitis B surface antigen |
| HCV | Hepatitis C virus |
| HDV | Hepatitis delta virus |
| HGF | Hepatocyte growth factor |
| HIV | Human immunodeficiency virus |
| HMGB1 | High mobility group box-1 |
| HPV | Human papilloma virus |
| HSP | Heat shock protein |
| HSV | Herpes simplex virus |
| ICP | Infected cell protein |
| IFIT1 | Interferon-induced protein with tetratricopeptide repeats 1 |
| IFI16 | IFN-gamma-inducible protein 16 |
| IFN | Interferon |
| IKK | Inhibitor of nuclear factor κb kinase |
| iNOS | Inducible nitric oxide synthetase |
| IRAK | IL-1R-associted kinase |
| IRF | Interferon regulatory factor |
| ISRE | Interferon-sensitive response element |
| JAK | Janus kinase |
| JNK | Jun N-terminal kinase |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| LPS | Lipopolysaccharide |
| LRRFIP1 | Leucine-rich repeat flightless-interacting protein 1 |
| LTR | Long terminal repeat |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral signaling protein |
| MDA5 | Melanoma differentiation-associated gene 5 |
| MDP | Muramyl dipeptide |
| MEF | Mouse embryonic fibroblasts |
| MHC | Major histocompatibility complex |
| MPL | Monophosphoryl lipid A |
| MyD88 | Myeloid differentiation protein 88 |
| NF-κB | Nuclear factor-κB |
| NLR | NOD-like receptor |
| NLRP3 | NACHT, LRR and PYD domain-containing protein 3 |
| CNS | Central nervous system |
| NO | Nitric oxide |
| NOD | Nucleotide-binding oligomerization domain |
| OAS | 2’-5’ oligoadenylate synthetase |
| ODN | Oligodeoxynucleotides |
| PAMP | Pathogen-associated molecular pattern |
| PBMC | Peripheral blood mononuclear cells |
| pDC | Plasmacytoid dendritic cells |
| PKR | Protein kinase R |
| PRR | Pathogen recognition receptor |
| PYHIN | Pyrin and HIN domain-containing protein |
| RLR | RIG-like receptor |
| RIG-I | Retinoic acid inducible gene I |
| RSV | Respiratory syncytial virus |
| RT | Reverse transcriptase |
| SAMHD1 | SAM domain and HD domain-containing protein 1 |
| SNP | Single-nucleotide polymorphism |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of IFN genes |
| TBK1 | TANK-binding kinase 1 |
| TDF | Tenofovir disproxyl fumerate |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor α |
| TRAF | TNF receptor-associated factor |
| TRIF | Toll/IL-1 receptor domain-containing adaptor inducing IFN-? |
| TRIM5α | Tripartite motif 5α |
| VAI | Adenoviral virus-associated type I |
| VSV | Vesicular stomatitis virus |
| VV | Vaccinia virus |
| VZV | Varicella zoster virus |
| WNV | West Nile virus |
1. Introduction
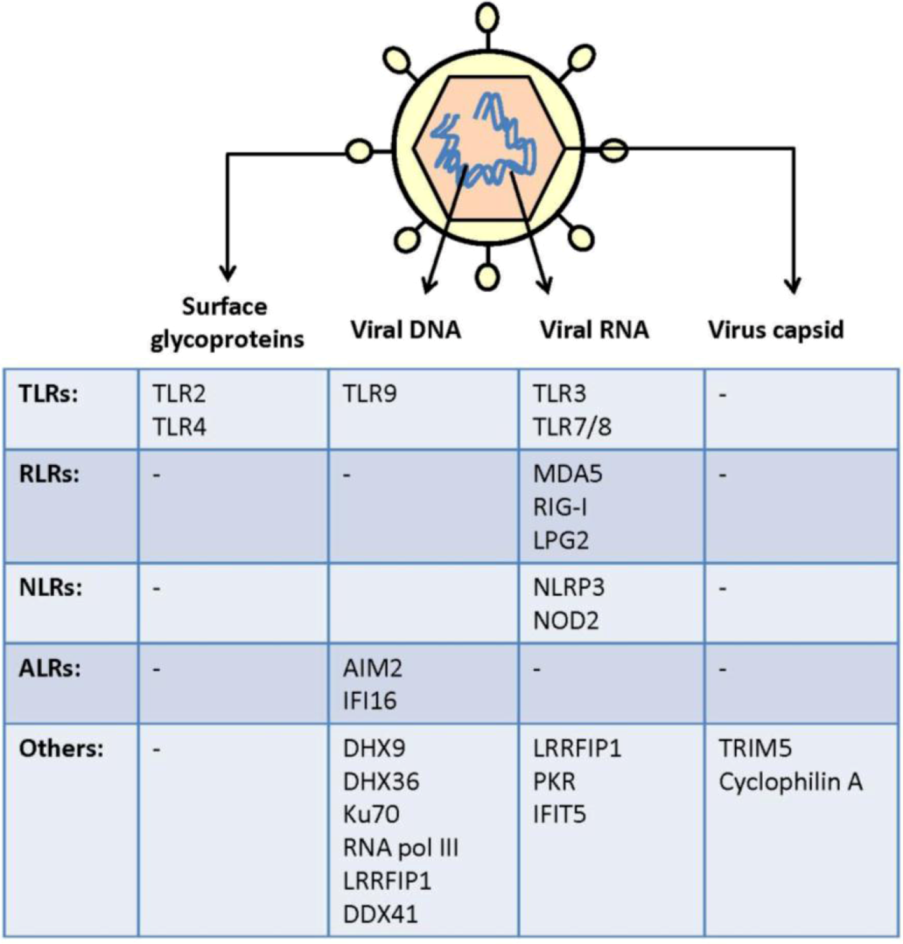
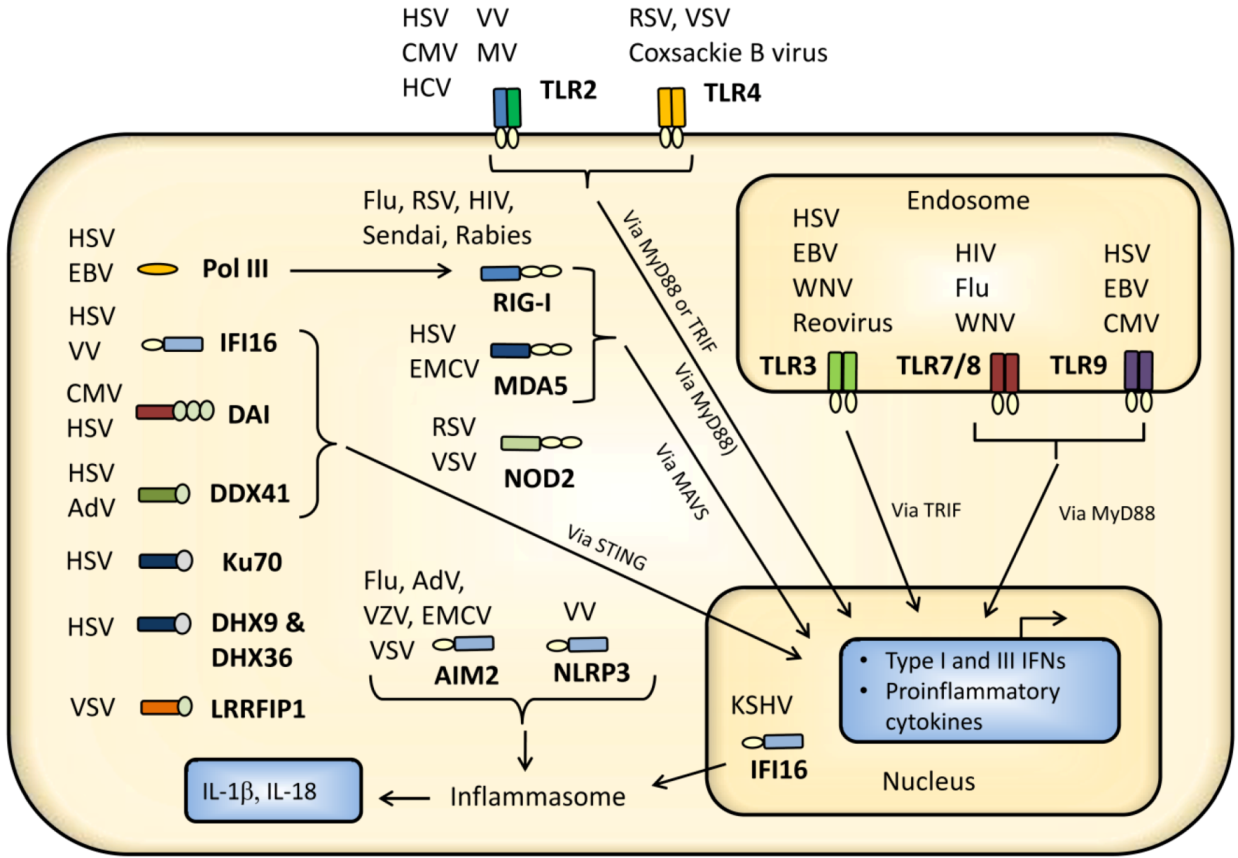
2. Virus Activation of Pattern Recognition Receptors


{kind=link}
{kind=link}
{kind=link}
| Viruses | Genome | Family | PAMPs | Primary host (s) |
|---|---|---|---|---|
| HSV | dsDNA | Herpesviridae | Glycoproteins, dsRNA, Viral DNA | Human |
| VZV | dsDNA | Herpesviridae | Glycoproteins, dsRNA, Viral DNA | Human |
| HCMV | dsDNA | Herpesviridae | Glycoproteins, dsRNA, Viral DNA | Human |
| EBV | dsDNA | Herpesviridae | Glycoproteins, Viral DNA, RNAs | Human |
| Vaccinia virus (VV) | dsDNA | Poxviridae | Glycoproteins, Viral DNA, RNAs | Unknown |
| Reovirus | dsRNA | Reoviridae | dsRNA genome | Human |
| Influenza A | (-)ssRNA | Orthomyxoviridae | Viral 5’ppp ssRNA | Human, Pig, Fowl |
| Measles virus | (-)ssRNA | Paramyxoviridae | dsRNA, surface hemaglutinin | Human |
| RSV | (-)ssRNA | Paramyxovirus | dsRNA, ssRNA, proteins | Human |
| Sendai virus | (-)ssRNA | Paramyxoviridae | dsRNA, ssRNA virus genome | Mouse |
| VSV | (-)ssRNA | Rhabdoviridae | RNA | Many |
| West Nile Virus | (+)ssRNA | Flaviviridea | Genomic RNA | Human |
| HCV | (+)ssRNA | Flaviviridae | RNA, NS protein | Human |
| Rhinovirus | (+)ssRNA | Picornaviridea | RNA | Human |
| Coxsackie virus | (+)ssRNA | Picornaviridae | Virion, dsRNA | Human |
| EMCV | (+)ssRNA | Picornaviridae | dsRNA | Pig, rodent |
| HIV | ssRNA (RT) | Retroviridae | Genomic RNA, cDNA, capsid, glycoproteins | Human |
2.1. Cell Surface Recognition of Virus

| Receptor | Virus PAMP | Virus | References |
|---|---|---|---|
| Cell surface TLRs | |||
| TLR2 | Glycoproteins gH/gL | HSV | [4,5] |
| Envelope glycoproteins | CMV | [6,7] | |
| Virion component, dUTase | EBV | [8,9] | |
| Not determined | VZV | [10] | |
| Hemagglutinin | Measles virus | [11] | |
| Core and nonstructural protein | HCV | [12] | |
| Not determined | VV | [13,14] | |
| TLR4 | Fusion protein | RSV | [15] |
| Not determined | Coxsackie virus B | [16] | |
| Glycoprotein | VSV | [17] | |
| Endosomally located TLRs | |||
| TLR3 | Virus-derived dsRNA | HSV | [18] |
| EBER RNA | EBV | [19] | |
| Genomic dsRNA | Reovirus | [20] | |
| RNA | Influenza virus | [21,22,23] | |
| dsRNA | RSV | [24,25] | |
| dsRNA | HIV-1 vector | [26] | |
| dsRNA | Rhinovirus | [27,28] | |
| RNA | WNV | [29,30] | |
| TLR7/8 | Genomic ssRNA | HIV | [31,32,33] |
| Genomic ssRNA | Influenza A | [34] | |
| Genomic ssRNA | Sendai | [35] | |
| Genomic ssRNA | Coxsackievirus B | [36] | |
| Genomic ssRNA | VSV | [34] | |
| TLR9 | Viral DNA | HSV | [37,38,39,40,41] |
| Viral DNA | CMV | [42] | |
| Viral DNA | VZV | [43] | |
| Viral DNA | EBV | [44,45] | |
| Viral DNA | KSHV | [46] | |
| Viral DNA | VV | [47] | |
| Viral DNA | Adenovirus | [48,49] | |
2.1.1. TLR2 and TLR4
2.1.2. C-Type Lectins
2.2. Endosomal Recognition of Viral RNA and DNA
2.2.1. TLR3
2.2.2. TLR7 and TLR8
2.2.3. TLR9
2.3. Cytoplasmic and Nuclear Recognition of Virus Infection
2.3.1. RIG-I-Like Receptors

| Receptor | Virus PAMP | Virus | References |
|---|---|---|---|
| Cytoplasmic RNA recognition | |||
| RIG-I | 5´ppp viral RNA | Influenza A | [23,90,91,92] |
| ssRNA and dsRNA | HIV | [93,94] | |
| Virus-encoded RNA | EBV | [95] | |
| dsRNA | Reovirus | [96,97] | |
| dsRNA | VV | [98] | |
| dsRNA | Measles virus | [99] | |
| RNA | RSV | [91,100] | |
| dsRNA | Sendai virus | [35,92,101] | |
| dsRNA | Human parainfluenza virus | [102] | |
| dsRNA | VSV | [92,101,103] | |
| MDA5 | dsRNA | HSV | [104] |
| dsRNA | VV | [98,105] | |
| RNA | Reovirus | [97] | |
| dsRNA | Measles Virus | [99] | |
| dsRNA | Coxsackie B | [106] | |
| RNA | Sendai virus defective interfering particles | [107] | |
| dsRNA | Rhinovirus | [27,28] | |
| dsRNA | EMCV | [92] | |
| DDX60 | dsRNA | VSV | [108] |
| DHX9 | dsRNA | Influenza A | [109] |
| dsRNA | Reovirus | [109] | |
| DDX1/DDX21/DHX36 | dsRNA | Influenza A | [110] |
| DDX1/DDX21/DHX36 | dsRNA | Reovirus | [110] |
| NOD2 | ssRNA | RSV | [111] |
| ssRNA | VSV | [111] | |
| NALP3 | M2 protein, RNA | Influenza | [112,113,114,115] |
| Unknown | Sendai virus | [112] | |
| dsRNA | EMCV | [116] | |
| dsRNA | VSV | [116] | |
| Genomic DNA | Adenovirus | [117] | |
| unknown | VZV | [118] | |
| PKR | dsRNA | HSV | [41,119] |
| dsRNA | VV | [98] | |
| LRRFIP1 | RNA | VSV | [120] |
| Cytoplasmic DNA recognition | |||
| RNA pol III | Genomic DNA | HSV | [121] |
| Genomic DNA | EBV | [122] | |
| IFI16 | Genomic DNA | HSV | [123] |
| DAI | Genomic DNA | HSV | [124] |
| Genomic DNA | CMV | [125] | |
| DHX9 | Genomic DNA | HSV | [126] |
| DHX36 | Genomic DNA | HSV | [126] |
| DDX41 | Genomic DNA | AdV | [127] |
| Genomic DNA | HSV | [127] | |
| Ku70 | Genomic DNA | HSV | [128] |
| AIM2 | Virus DNA | MCMV | [129] |
| Virus DNA | VV | [129] | |
| Nuclear-located receptor for nucleic acids | |||
| IFI16 | Virus genomic DNA | KSHV | [130] |
| Miscellaneous | |||
| Cyclophilin A | Capsid | HIV | [131] |
| TRIM5 | Capsid lattice | HIV | [132] |
| NLRP3 | Membrane penetration | AdV | [75] |
| IFIT1 | 5’triphosphated viral RNA | Influenza A | [133] |
2.3.2. Other DExD/H-Box Helicases
2.3.3. NOD-Like Receptors
2.3.4. PKR
2.3.5. DNA Receptors
2.3.6. DAI/ZBP-1
2.3.7. Ku70
2.3.8. IFI16
2.3.9. RNA pol III
2.3.10. DHX9, DHX36, DDX41, and DDX60
2.3.11. LRRFIP1
2.3.12. AIM2
2.4. Other Viral Sensors and Innate Mediators
2.4.1. Sensing of Viral Capsids
2.4.2. Membrane Fusion Events
2.4.3. HMBG1
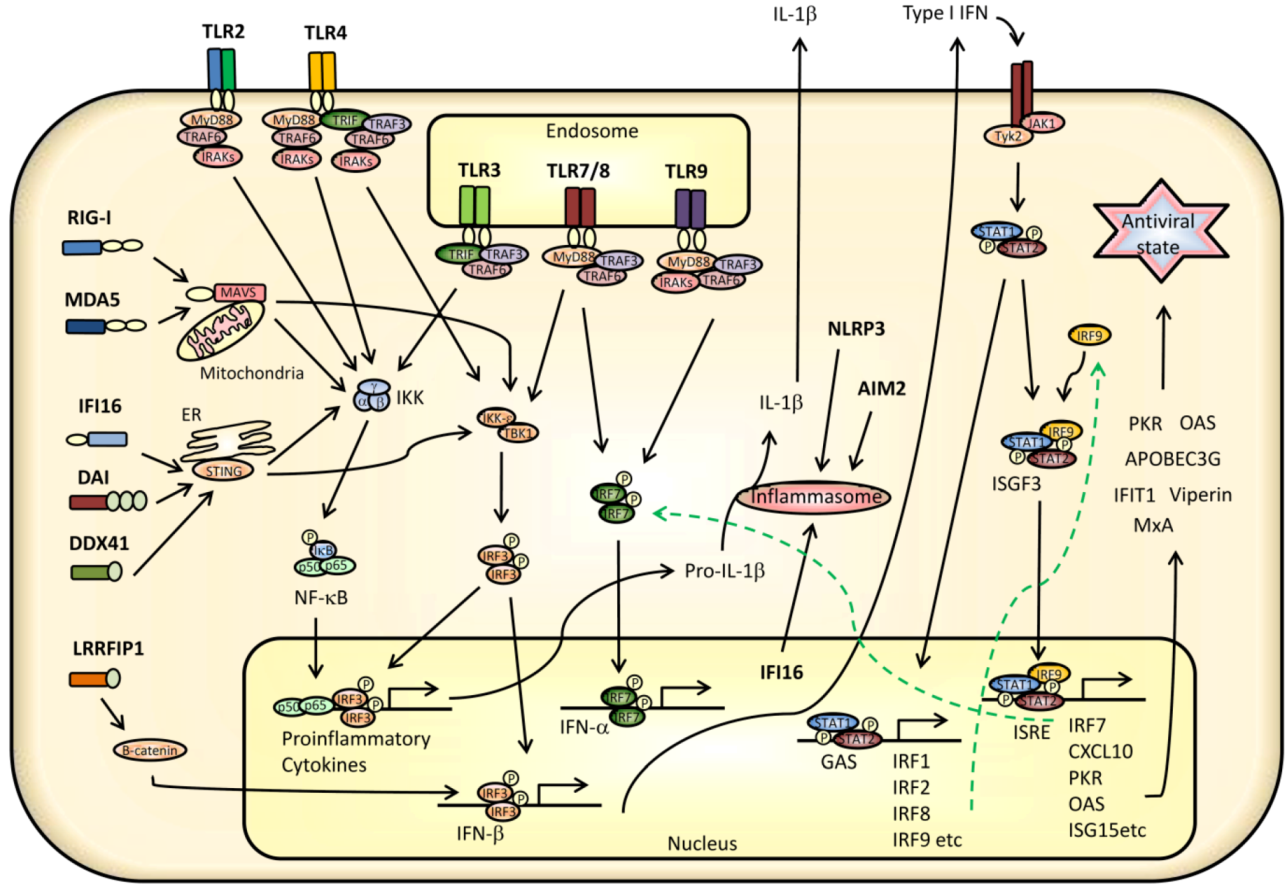
3. Innate Signaling Restricting Virus Infection
3.1. TLR Signaling
3.2. Signaling from RLRs and Cytoplasmic DNA Receptors
3.3. Transcriptional Regulation of IFNs, Cytokines, and ISGs
5. Therapeutic Implications of Innate Stimulation
5.1. TLR and NLR Agonists
5.1.1. TLR2 Agonists
5.1.2. TLR3 Agonists
5.1.3. TLR4 Agonists
5.1.4. TLR7/8 Agonists
5.1.5. TLR9 Agonists
5.1.6. NLR Agonists
5.2. Targeting Cytoplasmic DNA Receptors
6. Immunomodulatory Effect of Antimicrobial Drugs
7. Concluding Remarks
Conflict of Interest
Acknowledgements
References
- Ishii, K.J.; Koyama, S.; Nakagawa, A.; Coban, C.; Akira, S. Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe. 2008, 3, 352–363. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 1315–1320. [Google Scholar]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient To Activate NF-kappaB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.; Jones, C.; Williams, M.V. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar]
- Wang, J.P.; Kurt-Jones, E.A.; Shin, O.S.; Manchak, M.D.; Levin, M.J.; Finberg, R.W. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J. Virol. 2005, 79, 12658–12666. [Google Scholar]
- Bieback, K.; Lien, E.; Klagge, I.M.; et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Oak, S.; Kodys, K.; et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar]
- Zhu, J.; Martinez, J.; Huang, X.; Yang, Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 2007, 109, 619–625. [Google Scholar] [CrossRef]
- Martinez, J.; Huang, X.; Yang, Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog 2010, 6, e1000811. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Triantafilou, K.; Triantafilou, M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J. Virol. 2004, 78, 11313–11320. [Google Scholar] [CrossRef]
- Georgel, P.; Jiang, Z.; Kunz, S.; et al. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 2007, 362, 304–313. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar]
- Iwakiri, D.; Zhou, L.; Samanta, M.; et al. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll- like receptor 3. Nature 2001, 413, 732–738. [Google Scholar]
- Guillot, L.; Le Goffic, R.; Bloch, S.; et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2006, 2, e53. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; et al. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar]
- Rudd, B.D.; Burstein, E.; Duckett, C.S.; Li, X.; Lukacs, N.W. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 2005, 79, 3350–3357. [Google Scholar] [CrossRef]
- Rudd, B.D.; Smit, J.J.; Flavell, R.A.; et al. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 2006, 176, 1937–1942. [Google Scholar]
- Breckpot, K.; Escors, D.; Arce, F.; et al. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef]
- Wang, Q.; Miller, D.J.; Bowman, E.R.; et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog 2011, 7, e1002070. [Google Scholar] [CrossRef]
- Wang, Q.; Nagarkar, D.R.; Bowman, E.R.; et al. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 2009, 183, 6989–6997. [Google Scholar] [CrossRef]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M., Jr.; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005, 115, 3265–3275. [Google Scholar]
- Alter, G.; Suscovich, T.J.; Teigen, N.; et al. Single-stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J. Immunol. 2007, 178, 7658–7666. [Google Scholar]
- Meier, A.; Alter, G.; Frahm, N.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; et al. Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005, 7, 1117–11126. [Google Scholar] [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; et al. Type I IFN production during herpes simplex virus infection is controlled by cell-type specific viral recognition through TLR9, the MAVS pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17343–17348. [Google Scholar]
- Malmgaard, L.; Melchjorsen, J.; Bowie, A.G.; Mogensen, S.C.; Paludan, S.R. Viral activation of macrophages through TLR-dependent and -independent pathways. J. Immunol. 2004, 173, 6890–6898. [Google Scholar]
- Varani, S.; Cederarv, M.; Feld, S.; et al. Human cytomegalovirus differentially controls B cell and T cell responses through effects on plasmacytoid dendritic cells. J. Immunol. 2007, 179, 7767–7776. [Google Scholar]
- Yu, H.R.; Huang, H.C.; Kuo, H.C.; et al. IFN-alpha production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell Mol. Immunol. 2011, 8, 181–188. [Google Scholar] [CrossRef]
- Lim, W.H.; Kireta, S.; Russ, G.R.; Coates, P.T. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 2007, 109, 1043–1050. [Google Scholar]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef]
- West, J.A.; Gregory, S.M.; Sivaraman, V.; Su, L.; Damania, B. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J. Virol. 2011, 85, 895–904. [Google Scholar] [CrossRef]
- Samuelsson, C.; Hausmann, J.; Lauterbach, H.; et al. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J. Clin. Invest. 2008, 118, 1776–1784. [Google Scholar] [CrossRef]
- Basner-Tschakarjan, E.; Gaffal, E.; O'Keeffe, M.; et al. Adenovirus efficiently transduces plasmacytoid dendritic cells resulting in TLR9-dependent maturation and IFN-alpha production. J. Gene. Med. 2006, 8, 1300–1306. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; McBride, A.; et al. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar]
- Awomoyi, A.A.; Rallabhandi, P.; Pollin, T.I.; et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 2007, 179, 3171–3177. [Google Scholar]
- Hutchens, M.A.; Luker, K.E.; Sonstein, J.; Nunez, G.; Curtis, J.L.; Luker, G.D. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathog 2008, 4, e1000153. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Barbalat, R.; Lau, L.; Locksley, R.M.; Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 2009, 10, 1200–1207. [Google Scholar] [CrossRef]
- Kijpittayarit, S.; Eid, A.J.; Brown, R.A.; Paya, C.V.; Razonable, R.R. Relationship between Toll-like receptor 2 polymorphism and cytomegalovirus disease after liver transplantation. Clin. Infect. Dis. 2007, 44, 1315–1320. [Google Scholar] [CrossRef]
- Ahmad, R.; El, B.S.; Cordeiro, P.; Menezes, J. Requirement of TLR2-mediated signaling for the induction of IL-15 gene expression in human monocytic cells by HSV-1. Blood 2008, 112, 2360–2368. [Google Scholar] [CrossRef]
- Mansur, D.S.; Kroon, E.G.; Nogueira, M.L.; et al. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am. J. Pathol. 2005, 166, 1419–1426. [Google Scholar] [CrossRef]
- Bochud, P.Y.; Magaret, A.S.; Koelle, D.M.; Aderem, A.; Wald, A. Polymorphisms in TLR2 are associated with increased viral shedding and lesional rate in patients with genital herpes simplex virus Type 2 infection. J. Infect. Dis. 2007, 196, 505–509. [Google Scholar] [CrossRef]
- Sorensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J. Immunol. 2008, 181, 8604–8612. [Google Scholar]
- Reske, A.; Pollara, G.; Krummenacher, C.; Katz, D.R.; Chain, B.M. Glycoprotein-dependent and TLR2-independent innate immune recognition of herpes simplex virus-1 by dendritic cells. J. Immunol. 2008, 180, 7525–7536. [Google Scholar]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Chase, M.A.; Senft, A.P.; Poynter, S.E.; Wong, H.R.; Page, K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 2009, 10, 31. [Google Scholar] [CrossRef]
- Borde, C.; Barnay-Verdier, S.; Gaillard, C.; Hocini, H.; Marechal, V.; Gozlan, J. Stepwise release of biologically active HMGB1 during HSV-2 infection. PLoS ONE 2011, 6, e16145. [Google Scholar]
- Turville, S.G.; Santos, J.J.; Frank, I.; et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood 2004, 103, 2170–2179. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Arrighi, J.F.; Pion, M.; Garcia, E.; et al. DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J. Exp. Med. 2004, 200, 1279–1288. [Google Scholar] [CrossRef]
- Hodges, A.; Sharrocks, K.; Edelmann, M.; et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat. Immunol. 2007, 8, 569–577. [Google Scholar]
- Lai, J.; Bernhard, O.K.; Turville, S.G.; Harman, A.N.; Wilkinson, J.; Cunningham, A.L. Oligomerization of the macrophage mannose receptor enhances gp120-mediated binding of HIV-1. J. Biol. Chem. 2009, 284, 11027–11038. [Google Scholar]
- Gringhuis, S.I.; van d, V.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muniz, O.; Corbi, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef]
- Halary, F.; Amara, A.; Lortat-Jacob, H.; et al. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity 2002, 17, 653–664. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef]
- Soby, S.; Laursen, R.R.; Ostergaard, L.; Melchjorsen, J. HSV-1-induced chemokine expression via IFI16-dependent and IFI16-independent pathways in human monocyte-derived macrophages. Herpesviridae 2012, 3, 6. [Google Scholar] [CrossRef]
- Barlan, A.U.; Griffin, T.M.; McGuire, K.A.; Wiethoff, C.M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Reinert, L.S.; Harder, L.; Holm, C.K.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J. Clin. Invest. 2012, 122, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Hutchens, M.; Luker, K.E.; Sottile, P.; et al. TLR3 increases disease morbidity and mortality from vaccinia infection. J. Immunol. 2008, 180, 483–491. [Google Scholar]
- Schulz, O.; Diebold, S.S.; Chen, M.; et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Wang, J.P.; Liu, P.; Latz, E.; Golenbock, D.T.; Finberg, R.W.; Libraty, D.H. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J. Immunol. 2006, 177, 7114–7121. [Google Scholar]
- Martinez, J.; Huang, X.; Yang, Y. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6442–6447. [Google Scholar] [CrossRef]
- Bauer, S.; Bathke, B.; Lauterbach, H.; et al. A major role for TLR8 in the recognition of vaccinia viral DNA by murine pDC? Proc. Natl. Acad. Sci. U.S.A. 2010, 107, E139. [Google Scholar] [CrossRef]
- Lepelley, A.; Louis, S.; Sourisseau, M.; et al. Innate Sensing of HIV-Infected Cells. PLoS Pathog 2011, 7, e1001284. [Google Scholar] [CrossRef] [Green Version]
- Wagner, H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004, 25, 381–386. [Google Scholar] [CrossRef]
- Hochrein, H.; Schlatter, B., O'Keeffe; et al. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11416–11421. [Google Scholar]
- Megjugorac, N.J.; Young, H.A.; Amrute, S.B.; Olshalsky, S.L.; Fitzgerald-Bocarsly, P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J. Leukoc. Biol. 2004, 75, 504–514. [Google Scholar]
- Megjugorac, N.J.; Gallagher, G.E.; Gallagher, G. Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29). J. Leukoc. Biol. 2009, 86, 1359–1363. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef]
- Berg, R.K.; Melchjorsen, J.; Rintahaka, J.; et al. Genomic HIV RNA Induces Innate Immune Responses through RIG-I-Dependent Sensing of Secondary-Structured RNA. PLoS ONE 2012, 7, e29291. [Google Scholar]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO. J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; et al. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar]
- Sen, A.; Pruijssers, A.J.; Dermody, T.S.; Garcia-Sastre, A.; Greenberg, H.B. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 2011, 85, 3717–3732. [Google Scholar] [CrossRef]
- Myskiw, C.; Arsenio, J.; Booy, E.P.; et al. RNA species generated in vaccinia virus infected cells activate cell type-specific MDA5 or RIG-I dependent interferon gene transcription and PKR dependent apoptosis. Virology 2011, 413, 183–193. [Google Scholar] [CrossRef]
- Ikegame, S.; Takeda, M.; Ohno, S.; Nakatsu, Y.; Nakanishi, Y.; Yanagi, Y. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 2010, 84, 372–379. [Google Scholar] [CrossRef]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef]
- Kato, H.; Sato, S.; Yoneyama, M.; et al. Cell Type-Specific Involvement of RIG-I in Antiviral Response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef]
- Sabbah, A.; Bose, S. Retinoic acid inducible gene I activates innate antiviral response against human parainfluenza virus type 3. Virol. J. 2009, 6, 200. [Google Scholar] [CrossRef]
- Poeck, H.; Bscheider, M.; Gross, O.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Rintahaka, J.; Soby, S.; et al. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; et al. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef]
- Yount, J.S.; Gitlin, L.; Moran, T.M.; Lopez, C.B. MDA5 Participates in the Detection of Paramyxovirus Infection and Is Essential for the Early Activation of Dendritic Cells in Response to Sendai Virus Defective Interfering Particles. J. Immunol. 2008, 180, 4910–4918. [Google Scholar]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. DDX60, a DExD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Lu, N.; Facchinetti, V.; Liu, Y.J. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol. 2011, 187, 4501–4508. [Google Scholar] [CrossRef]
- Zhang, Z.; Kim, T.; Bao, M.; et al. DDX1, DDX21, and DHX36 Helicases Form a Complex with the Adaptor Molecule TRIF to Sense dsRNA in Dendritic Cells. Immunity 2011, 34, 866–878. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; et al. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Rajan, J.V.; Rodriguez, D.; Miao, E.A.; Aderem, A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 2011, 85, 4167–4172. [Google Scholar] [CrossRef]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar]
- Nour, A.M.; Reichelt, M.; Ku, C.C.; Ho, M.Y.; Heineman, T.C.; Arvin, A.M. Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J. Biol. Chem. 2011, 286, 17921–17933. [Google Scholar]
- Melchjorsen, J.; Pedersen, F.S.; Mogensen, S.C.; Paludan, S.R. Herpes simplex virus selectively induces expression of the CC Chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J. Virol. 2002, 76, 2780–2788. [Google Scholar] [CrossRef]
- Yang, P.; An, H.; Liu, X.; et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 2010, 11, 487–494. [Google Scholar]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Fruh, K. Human Cytomegalovirus Induces the Interferon Response Via the DNA Sensor ZBP1. J. Virol. 2009, 84, 585–598. [Google Scholar]
- Kim, T.; Pazhoor, S.; Bao, M.; et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15181–15186. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar]
- Zhang, X.; Brann, T.W.; Zhou, M.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; et al. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar]
- Pertel, T.; Hausmann, S.; Morger, D.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; et al. IFIT1 is an antiviral protein that recognizes 5'-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar]
- Li, K.; Chen, Z.; Kato, N.; Gale, M., Jr.; Lemon, S.M. Distinct poly-I: C and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-kappaB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-beta Signaling. Mol. Cell. 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Venkataraman, T.; Valdes, M.; Elsby, R.; et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; et al. The RNA Helicase Lgp2 Inhibits TLR-Independent Sensing of Viral Replication by Retinoic Acid-Inducible Gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar]
- Satoh, T.; Kato, H.; Kumagai, Y.; et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1512–1517. [Google Scholar]
- Broquet, A.H.; Hirata, Y.; McAllister, C.S.; Kagnoff, M.F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 2011, 186, 1618–1626. [Google Scholar] [CrossRef]
- Burdette, D.; Haskett, A.; Presser, L.; McRae, S.; Iqbal, J.; Waris, G. Hepatitis C virus activates interleukin-1beta via caspase-1-inflammasome complex. J. Gen. Virol. 2012, 93, 235–246. [Google Scholar] [CrossRef]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; et al. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar]
- Pontillo, A.; Silva, L.T.; Oshiro, T.M.; Finazzo, C.; Crovella, S.; Duarte, A.J. HIV-1 induces NALP3-inflammasome expression and interleukin-1beta secretion in dendritic cells from healthy individuals but not from HIV-positive patients. AIDS 2012, 26, 11–18. [Google Scholar] [CrossRef]
- Pontillo, A.; Brandao, L.A.; Guimaraes, R.L.; Segat, L.; Athanasakis, E.; Crovella, S. A 3'UTR SNP in NLRP3 gene is associated with susceptibility to HIV-1 infection. J. Acquir. Immune. Defic. Syndr. 2010, 54, 236–240. [Google Scholar] [CrossRef]
- Pontillo, A.; Oshiro, T.M.; Girardelli, M.; Kamada, A.J.; Crovella, S.; Duarte, A.J. Polymorphisms in inflammasome' genes and susceptibility to HIV-1 infection. J. Acquir. Immune. Defic. Syndr. 2012, 59, 121–125. [Google Scholar] [CrossRef]
- Suzuki, K.; Mori, A.; Ishii, K.J.; et al. Activation of target-tissue immune-recognition molecules by double-stranded polynucleotides. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2285–2290. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Furr, S.R.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Marriott, I. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflammation 2011, 8, 99. [Google Scholar] [CrossRef]
- Hayashi, T.; Nishitsuji, H.; Takamori, A.; Hasegawa, A.; Masuda, T.; Kannagi, M. DNA-dependent activator of IFN-regulatory factors enhances the transcription of HIV-1 through NF-kappaB. Microbes. Infect. 2010, 12, 937–947. [Google Scholar] [CrossRef]
- Downs, J.A.; Jackson, S.P. A means to a DNA end: the many roles of Ku. Nat. Rev. Mol. Cell. Biol. 2004, 5, 367–378. [Google Scholar] [CrossRef]
- Stein, S.C.; Falck-Pedersen, E. Sensing adenovirus infection: activation of interferon regulatory factor 3 in RAW 264.7 cells. J. Virol. 2012, 86, 4527–4537. [Google Scholar] [CrossRef]
- Veeranki, S.; Duan, X.; Panchanathan, R.; Liu, H.; Choubey, D. IFI16 Protein Mediates the Anti-inflammatory Actions of the Type-I Interferons through Suppression of Activation of Caspase-1 by Inflammasomes. PLoS ONE 2011, 6, e27040. [Google Scholar]
- Gariano, G.R.; Dell'Oste, V.; Bronzini, M.; et al. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 2012, 8, e1002498. [Google Scholar] [CrossRef]
- Conrady, C.D.; Zheng, M.; Fitzgerald, K.A.; Liu, C.; Carr, D.J. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal. Immunol. 2012, 5, 173–183. [Google Scholar] [CrossRef]
- Kis-Toth, K.; Szanto, A.; Thai, T.H.; Tsokos, G.C. Cytosolic DNA-Activated Human Dendritic Cells Are Potent Activators of the Adaptive Immune Response. J. Immunol. 2011, 187, 1222–1234. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kawabata, K.; Kouyama, E.; et al. Induction of type I interferon by adenovirus-encoded small RNAs. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17286–17291. [Google Scholar] [CrossRef]
- Dai, P.; Jeong, S.Y.; Yu, Y.; et al. Modulation of TLR signaling by multiple MyD88-interacting partners including leucine-rich repeat Fli-I-interacting proteins. J. Immunol. 2009, 182, 3450–3460. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Zhou, X.; Rajaiya, J.; Chodosh, J. Viral capsid is a pathogen-associated molecular pattern in adenovirus keratitis. PLoS Pathog 2010, 6, e1000841. [Google Scholar] [CrossRef]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 2001, re2. [Google Scholar]
- Nallagatla, S.R.; Toroney, R.; Bevilacqua, P.C. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr. Opin. Struct. Biol. 2011, 21, 119–127. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef]
- Nallagatla, S.R.; Hwang, J.; Toroney, R.; Zheng, X.; Cameron, C.E.; Bevilacqua, P.C. 5'-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 2007, 318, 1455–1458. [Google Scholar] [CrossRef]
- Kim, I.; Liu, C.W.; Puglisi, J.D. Specific recognition of HIV TAR RNA by the dsRNA binding domains (dsRBD1-dsRBD2) of PKR. J. Mol. Biol. 2006, 358, 430–442. [Google Scholar] [CrossRef]
- Arnaud, N.; Dabo, S.; Maillard, P.; et al. Hepatitis C virus controls interferon production through PKR activation. PLoS ONE 2010, 5, e10575. [Google Scholar] [CrossRef]
- Talloczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar]
- English, L.; Chemali, M.; Duron, J.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- McAllister, C.S.; Samuel, C.E. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 2009, 284, 1644–1651. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Yanai, H.; Ban, T.; Wang, Z.; et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar]
- Moisy, D.; Avilov, S.V.; Jacob, Y.; et al. HMGB1 protein binds to influenza virus nucleoprotein and promotes viral replication. J. Virol. 2012, 86, 9122–9133. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayashi, Y.; Omori, H.; et al. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 2012, 11, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Saidi, H.; Melki, M.T.; Gougeon, M.L. HMGB1-dependent triggering of HIV-1 replication and persistence in dendritic cells as a consequence of NK-DC cross-talk. PLoS ONE 2008, 3, e3601. [Google Scholar] [CrossRef]
- Thierry, S.; Gozlan, J.; Jaulmes, A.; et al. High-mobility group box 1 protein induces HIV-1 expression from persistently infected cells. AIDS 2007, 21, 283–292. [Google Scholar] [CrossRef]
- Cassetta, L.; Fortunato, O.; Adduce, L.; et al. Extracellular high mobility group box-1 inhibits R5 and X4 HIV-1 strains replication in mononuclear phagocytes without induction of chemokines and cytokines. AIDS 2009, 23, 567–577. [Google Scholar]
- Barqasho, B.; Nowak, P.; Abdurahman, S.; Walther-Jallow, L.; Sonnerborg, A. Implications of the release of high-mobility group box 1 protein from dying cells during human immunodeficiency virus type 1 infection in vitro. J. Gen. Virol. 2010, 91, 1800–1809. [Google Scholar] [CrossRef]
- Nowak, P.; Barqasho, B.; Sonnerborg, A. Elevated plasma levels of high mobility group box protein 1 in patients with HIV-1 infection. AIDS 2007, 21, 869–871. [Google Scholar] [CrossRef]
- Troseid, M.; Nowak, P.; Nystrom, J.; Lindkvist, A.; Abdurahman, S.; Sonnerborg, A. Elevated plasma levels of lipopolysaccharide and high mobility group box-1 protein are associated with high viral load in HIV-1 infection: reduction by 2-year antiretroviral therapy. AIDS 2010, 24, 1733–1737. [Google Scholar] [CrossRef]
- Yoneyama, M.; Fujita, T. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 2010, 20, 4–22. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Innate immune sensing of DNA viruses. Virology 2011, 411, 153–162. [Google Scholar] [CrossRef]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar]
- Lin, R.; Yang, L.; Nakhaei, P.; et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J. Biol. Chem. 2006, 281, 2095–2103. [Google Scholar]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Parker, D.; Martin, F.J.; Soong, G.; et al. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2011, 2, e00016–11. [Google Scholar]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Tsuruta, L.; Arai, N.; Arai, K. Transcriptional control of cytokine genes. Int. Rev. Immunol. 1998, 16, 581–616. [Google Scholar]
- Melchjorsen, J.; Sorensen, L.N.; Paludan, S.R. Expression and function of chemokines during viral infections: from molecular mechanisms to in vivo function. J. Leukoc. Biol. 2003, 74, 331–343. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Paludan, S.R. Induction of RANTES/CCL5 by herpes simplex virus is regulated by NF-kappaB and IRF3. J. Gen. Virol. 2003, 84, 2491–2495. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Kristiansen, H.; Christiansen, R.; et al. Differential Regulation of the OASL and OAS1 Genes in Response to Viral Infections. J. Interferon Cytokine Res. 2009, 29, 199–207. [Google Scholar] [CrossRef]
- Leonard, W.J.; O'Shea, J.J. Jaks and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–668. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Meager, A.; Visvalingam, K.; Dilger, P.; Bryan, D.; Wadhwa, M. Biological activity of interleukins-28 and -29: Comparison with type I interferons. Cytokine 2005, 31, 109–118. [Google Scholar] [CrossRef]
- Dellgren, C.; Gad, H.H.; Hamming, O.J.; Melchjorsen, J.; Hartmann, R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes Immun. 2009, 10, 125–131. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN-{lambda}), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In Vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef]
- Osterlund, P.; Veckman, V.; Siren, J.; et al. Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J. Virol. 2005, 79, 9608–9617. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Siren, J.; Julkunen, I.; Paludan, S.R.; Matikainen, S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen. Virol. 2006, 87, 1099–1108. [Google Scholar] [CrossRef]
- Berghall, H.; Siren, J.; Sarkar, D.; et al. The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes Infect. 2006, 8, 2138–2144. [Google Scholar] [CrossRef]
- Coccia, E.M.; Severa, M.; Giacomini, E.; et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef]
- Hou, W.; Wang, X.; Ye, L.; et al. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J. Virol. 2009, 83, 3834–3842. [Google Scholar] [CrossRef]
- Robek, M.D.; Boyd, B.S.; Chisari, F.V. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 2005, 79, 3851–3854. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; et al. An important role for type III Interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar]
- Kerr, I.M.; Brown, R.E.; Hovanessian, A.G. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature 1977, 268, 540–542. [Google Scholar] [CrossRef]
- Floyd-Smith, G.; Slattery, E.; Lengyel, P. Interferon action: RNA cleavage pattern of a (2'-5')oligoadenylate--dependent endonuclease. Science 1981, 212, 1030–1032. [Google Scholar]
- Hovanessian, A.G. Interferon-induced and double-stranded RNA-activated enzymes: A specific protein kinase and 2',5'-oligoadenylate synthetases. J. Interferon. Res. 1991, 11, 199–205. [Google Scholar] [CrossRef]
- Rebouillat, D.; Marie, I.; Hovanessian, A.G. Molecular cloning and characterization of two related and interferon-induced 56-kDa and 30-kDa proteins highly similar to 2'-5' oligoadenylate synthetase. Eur. J. Biochem. 1998, 257, 319–330. [Google Scholar]
- Sharp, T.V.; Raine, D.A.; Gewert, D.R.; Joshi, B.; Jagus, R.; Clemens, M.J. Activation of the interferon-inducible (2'-5') oligoadenylate synthetase by the Epstein-Barr virus RNA, EBER-1. Virology 1999, 257, 303–313. [Google Scholar]
- Desai, S.Y.; Patel, R.C.; Sen, G.C.; Malhotra, P.; Ghadge, G.D.; Thimmapaya, B. Activation of interferon-inducible 2'-5' oligoadenylate synthetase by adenoviral VAI RNA. J. Biol. Chem. 1995, 270, 3454–3461. [Google Scholar]
- Maitra, R.K.; McMillan, N.A.; Desai, S.; et al. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology 1994, 204, 823–827. [Google Scholar] [CrossRef]
- Yakub, I.; Lillibridge, K.M.; Moran, A.; et al. Single nucleotide polymorphisms in genes for 2'-5'-oligoadenylate synthetase and RNase L inpatients hospitalized with West Nile virus infection. J. Infect. Dis. 2005, 192, 1741–1748. [Google Scholar] [CrossRef]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Rios, J.J.; Fleming, J.G.; Bryant, U.K.; et al. OAS1 polymorphisms are associated with susceptibility to West Nile encephalitis in horses. PLoS ONE 2010, 5, e10537. [Google Scholar]
- Knapp, S.; Yee, L.J.; Frodsham, A.J.; et al. Polymorphisms in interferon-induced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003, 4, 411–419. [Google Scholar] [CrossRef]
- El Awady, M.K.; Anany, M.A.; Esmat, G.; et al. Single nucleotide polymorphism at exon 7 splice acceptor site of OAS1 gene determines response of hepatitis C virus patients to interferon therapy. J. Gastroenterol. Hepatol. 2011, 26, 843–850. [Google Scholar] [CrossRef]
- Haralambieva, I.H.; Dhiman, N.; Ovsyannikova, I.G.; et al. 2'-5'-Oligoadenylate synthetase single-nucleotide polymorphisms and haplotypes are associated with variations in immune responses to rubella vaccine. Hum. Immunol. 2010, 71, 383–391. [Google Scholar] [CrossRef]
- Kristiansen, H.; Scherer, C.A.; McVean, M.; et al. Extracellular 2'-5' oligoadenylate synthetase stimulates RNase L-independent antiviral activity: a novel mechanism of virus-induced innate immunity. J. Virol. 2010, 84, 11898–11904. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1371–1376. [Google Scholar] [CrossRef]
- Pincetic, A.; Kuang, Z.; Seo, E.J.; Leis, J. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 2010, 84, 4725–4736. [Google Scholar] [CrossRef]
- Kuang, Z.; Seo, E.J.; Leis, J. Mechanism of Inhibition of Retrovirus Release from Cells by Interferon-Induced Gene ISG15. J. Virol. 2011, 85, 7153–7161. [Google Scholar] [CrossRef]
- Zhao, C.; Hsiang, T.Y.; Kuo, R.L.; Krug, R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2253–2258. [Google Scholar]
- Dai, J.; Pan, W.; Wang, P. ISG15 facilitates cellular antiviral response to dengue and west nile virus infection in vitro. Virol. J. 2011, 8, 468. [Google Scholar] [CrossRef]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 1440–1445. [Google Scholar] [CrossRef]
- Lu, G.; Reinert, J.T.; Pitha-Rowe, I.; et al. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol. Biol. 2006, 52, 29–41. [Google Scholar]
- Osiak, A.; Utermohlen, O.; Niendorf, S.; Horak, I.; Knobeloch, K.P. ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus. Mol. Cell. Biol. 2005, 25, 6338–6345. [Google Scholar] [CrossRef]
- Chieux, V.; Chehadeh, W.; Harvey, J.; Haller, O.; Wattre, P.; Hober, D. Inhibition of coxsackievirus B4 replication in stably transfected cells expressing human MxA protein. Virology 2001, 283, 84–92. [Google Scholar] [CrossRef]
- Gordien, E.; Rosmorduc, O.; Peltekian, C.; Garreau, F.; Brechot, C.; Kremsdorf, D. Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J. Virol. 2001, 75, 2684–2691. [Google Scholar] [CrossRef]
- Turan, K.; Mibayashi, M.; Sugiyama, K.; Saito, S.; Numajiri, A.; Nagata, K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic. Acids. Res. 2004, 32, 643–652. [Google Scholar] [CrossRef]
- Kochs, G.; Haller, O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2082–2086. [Google Scholar] [CrossRef]
- Kochs, G.; Janzen, C.; Hohenberg, H.; Haller, O. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3153–3158. [Google Scholar] [CrossRef]
- Hijikata, M.; Ohta, Y.; Mishiro, S. Identification of a single nucleotide polymorphism in the MxA gene promoter (G/T at nt -88) correlated with the response of hepatitis C patients to interferon. Intervirology 2000, 43, 124–127. [Google Scholar] [CrossRef]
- Suzuki, F.; Arase, Y.; Suzuki, Y.; et al. Single nucleotide polymorphism of the MxA gene promoter influences the response to interferon monotherapy in patients with hepatitis C viral infection. J. Viral. Hepat. 2004, 11, 271–276. [Google Scholar] [CrossRef]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 15125–15130. [Google Scholar] [CrossRef]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007, 2, 96–105. [Google Scholar] [CrossRef]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; et al. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef]
- Wang, S.; Wu, X.; Pan, T.; et al. Viperin inhibits hepatitis C virus replication by interfering with binding of NS5A to host protein hVAP-33. J. Gen. Virol. 2012, 93, 83–92. [Google Scholar] [CrossRef]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef]
- Lim, E.S.; Wu, L.I.; Malik, H.S.; Emerman, M. The function and evolution of the restriction factor viperin in primates was not driven by lentiviruses. Retrovirology 2012, 9, 55. [Google Scholar] [CrossRef]
- Saitoh, T.; Satoh, T.; Yamamoto, N.; et al. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 2011, 34, 352–363. [Google Scholar] [CrossRef]
- Fensterl, V.; Wetzel, J.L.; Ramachandran, S.; et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 2012, 8, e1002712. [Google Scholar] [CrossRef]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar]
- Wang, F.X.; Huang, J.; Zhang, H.; Ma, X.; Zhang, H. APOBEC3G upregulation by alpha interferon restricts human immunodeficiency virus type 1 infection in human peripheral plasmacytoid dendritic cells. J. Gen. Virol. 2008, 89, 722–730. [Google Scholar] [CrossRef]
- Argyris, E.G.; Acheampong, E.; Wang, F.; et al. The interferon-induced expression of APOBEC3G in human blood-brain barrier exerts a potent intrinsic immunity to block HIV-1 entry to central nervous system. Virology 2007, 367, 440–451. [Google Scholar] [CrossRef]
- Suspene, R.; Aynaud, M.M.; Koch, S.; et al. Genetic Editing of Herpes Simplex Virus 1 and Epstein-Barr Herpesvirus Genomes by Human APOBEC3 Cytidine Deaminases in Culture and In Vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 8321–8326. [Google Scholar] [CrossRef]
- Fehrholz, M.; Kendl, S.; Prifert, C.; et al. The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J. Gen. Virol. 2012, 93, 565–576. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef]
- Roa, A.; Hayashi, F.; Yang, Y.; et al. RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 2012, 86, 1717–1727. [Google Scholar] [CrossRef]
- Liu, F.L.; Qiu, Y.Q.; Li, H.; et al. An HIV-1 resistance polymorphism in TRIM5alpha gene among Chinese intravenous drug users. J. Acquir. Immune. Defic. Syndr. 2011, 56, 306–311. [Google Scholar]
- Price, H.; Lacap, P.; Tuff, J.; et al. A TRIM5alpha exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS 2010, 24, 1813–1821. [Google Scholar] [CrossRef]
- Javanbakht, H.; An, P.; Gold, B.; et al. Effects of human TRIM5alpha polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology 2006, 354, 15–27. [Google Scholar] [CrossRef]
- Le, T.A.; Willey, S.; Neil, S.J. Antiviral inhibition of enveloped virus release by tetherin/BST-2: Action and counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef]
- Kuhl, B.D.; Cheng, V.; Wainberg, M.A.; Liang, C. Tetherin and its viral antagonists. J. Neuroimmune. Pharmacol. 2011, 6, 188–201. [Google Scholar] [CrossRef]
- Bego, M.G.; Mercier, J.; Cohen, E.A. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J. Virol. 2012, 86, 3513–3527. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Barrett, B.S.; Smith, D.S.; Li, S.X.; Guo, K.; Hasenkrug, K.J.; Santiago, M.L. A single nucleotide polymorphism in tetherin promotes retrovirus restriction in vivo. PLoS Pathog 2012, 8, e1002596. [Google Scholar] [CrossRef]
- Kuhl, B.D.; Sloan, R.D.; Donahue, D.A.; Bar-Magen, T.; Liang, C.; Wainberg, M.A. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 2010, 7, 115. [Google Scholar] [CrossRef]
- Dafa-Berger, A.; Kuzmina, A.; Fassler, M.; Yitzhak-Asraf, H.; Shemer-Avni, Y.; Taube, R. Modulation of hepatitis C virus release by the interferon-induced protein BST-2/tetherin. Virology 2012, 428, 98–111. [Google Scholar] [CrossRef]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 2009, 83, 9672–9681. [Google Scholar] [CrossRef]
- Yasuda, J. Ebolavirus Replication and Tetherin/BST-2. Front Microbiol. 2012, 3, 111. [Google Scholar]
- Mangeat, B.; Cavagliotti, L.; Lehmann, M.; et al. Influenza Virus Partially Counteracts Restriction Imposed by Tetherin/BST-2. J. Biol. Chem. 2012, 287, 22015–22029. [Google Scholar] [CrossRef]
- Viswanathan, K.; Smith, M.S.; Malouli, D.; Mansouri, M.; Nelson, J.A.; Fruh, K. BST2/Tetherin enhances entry of human cytomegalovirus. PLoS Pathog 2011, 7, e1002332. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Tissot, C.; Mechti, N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem. 1995, 270, 14891–14898. [Google Scholar] [CrossRef]
- Bouazzaoui, A.; Kreutz, M.; Eisert, V.; et al. Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology 2006, 356, 79–94. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Marelli, S.S.; Pultrone, C.; et al. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 2011, 85, 5183–5196. [Google Scholar]
- Barr, S.D.; Smiley, J.R.; Bushman, F.D. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog 2008, 4, e1000007. [Google Scholar] [CrossRef]
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 2009, 50, 424–433. [Google Scholar]
- Eldin, P.; Papon, L.; Oteiza, A.; Brocchi, E.; Lawson, T.G.; Mechti, N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 2009, 90, 536–545. [Google Scholar]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Biswas, N.; Wang, T.; Ding, M.; et al. ADAR1 is a novel multi targeted anti-HIV-1 cellular protein. Virology 2012, 422, 265–277. [Google Scholar] [CrossRef]
- Taylor, D.R.; Puig, M.; Darnell, M.E.; Mihalik, K.; Feinstone, S.M. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005, 79, 6291–6298. [Google Scholar]
- Jayan, G.C.; Casey, J.L. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J. Virol. 2002, 76, 12399–12404. [Google Scholar] [CrossRef]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 15118–15123. [Google Scholar]
- Hartwig, D.; Schutte, C.; Warnecke, J.; et al. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J. Viral. Hepat. 2006, 13, 150–157. [Google Scholar] [CrossRef]
- Doria, M.; Tomaselli, S.; Neri, F.; et al. ADAR2 editing enzyme is a novel human immunodeficiency virus-1 proviral factor. J. Gen. Virol. 2011, 92, 1228–1232. [Google Scholar] [CrossRef]
- Clerzius, G.; Gelinas, J.F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Phuphuakrat, A.; Kraiwong, R.; Boonarkart, C.; Lauhakirti, D.; Lee, T.H.; Auewarakul, P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J. Virol. 2008, 82, 10864–10872. [Google Scholar] [CrossRef]
- Nie, Y.; Hammond, G.L.; Yang, J.H. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 2007, 81, 917–923. [Google Scholar] [CrossRef]
- Li, Z.; Wolff, K.C.; Samuel, C.E. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 2010, 396, 316–322. [Google Scholar] [CrossRef]
- Li, Z.; Okonski, K.M.; Samuel, C.E. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J. Virol. 2012, 86, 3787–3794. [Google Scholar] [CrossRef]
- Toth, A.M.; Li, Z.; Cattaneo, R.; Samuel, C.E. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 2009, 284, 29350–29356. [Google Scholar] [CrossRef]
- Rose, W.A.; McGowin, C.L.; Pyles, R.B. FSL-1, a bacterial-derived toll-like receptor 2/6 agonist, enhances resistance to experimental HSV-2 infection. Virol. J. 2009, 6, 195. [Google Scholar] [CrossRef]
- Gill, N.; Deacon, P.M.; Lichty, B.; Mossman, K.L.; Ashkar, A.A. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J. Virol. 2006, 80, 9943–9950. [Google Scholar] [CrossRef]
- Thibault, S.; Tardif, M.R.; Barat, C.; Tremblay, M.J. TLR2 signaling renders quiescent naive and memory CD4+ T cells more susceptible to productive infection with X4 and R5 HIV-type 1. J. Immunol. 2007, 179, 4357–4366. [Google Scholar]
- de Jong, M.A.; de Witte, L.; Oudhoff, M.J.; Gringhuis, S.I.; Gallay, P.; Geijtenbeek, T.B. TNF-alpha and TLR agonists increase susceptibility to HIV-1 transmission by human Langerhans cells ex vivo. J. Clin. Invest. 2008, 118, 3440–3452. [Google Scholar] [CrossRef]
- Thibault, S.; Fromentin, R.; Tardif, M.R.; Tremblay, M.J. TLR2 and TLR4 triggering exerts contrasting effects with regard to HIV-1 infection of human dendritic cells and subsequent virus transfer to CD4+ T cells. Retrovirology 2009, 6, 42. [Google Scholar] [CrossRef]
- Datta, S.K.; Redecke, V.; Prilliman, K.R.; et al. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J. Immunol. 2003, 170, 4102–4110. [Google Scholar]
- Weck, M.M.; Grunebach, F.; Werth, D.; Sinzger, C.; Bringmann, A.; Brossart, P. TLR ligands differentially affect uptake and presentation of cellular antigens. Blood 2007, 109, 3890–3894. [Google Scholar] [CrossRef]
- Suh, H.S.; Zhao, M.L.; Choi, N.; Belbin, T.J.; Brosnan, C.F.; Lee, S.C. TLR3 and TLR4 are innate antiviral immune receptors in human microglia: Role of IRF3 in modulating antiviral and inflammatory response in the CNS. Virology 2009, 392, 246–259. [Google Scholar] [CrossRef]
- Rivieccio, M.A.; Suh, H.S.; Zhao, Y.; et al. TLR3 Ligation Activates an Antiviral Response in Human Fetal Astrocytes: A Role for Viperin/cig5. J. Immunol. 2006, 177, 4735–4741. [Google Scholar]
- Brichacek, B.; Vanpouille, C.; Kiselyeva, Y.; et al. Contrasting roles for TLR ligands in HIV-1 pathogenesis. PLoS ONE 2010, 5, e12831. [Google Scholar]
- Herbst-Kralovetz, M.M.; Pyles, R.B. Quantification of poly(I:C)-mediated protection against genital herpes simplex virus type 2 infection. J. Virol. 2006, 80, 9988–9997. [Google Scholar] [CrossRef]
- Nazli, A.; Yao, X.D.; Smieja, M.; Rosenthal, K.L.; Ashkar, A.A.; Kaushic, C. Differential induction of innate anti-viral responses by TLR ligands against Herpes simplex virus, type 2, infection in primary genital epithelium of women. Antiviral. Res. 2009, 81, 103–112. [Google Scholar]
- MacDonald, E.M.; Savoy, A.; Gillgrass, A.; et al. Susceptibility of human female primary genital epithelial cells to herpes simplex virus, type-2 and the effect of TLR3 ligand and sex hormones on infection. Biol. Reprod. 2007, 77, 1049–1059. [Google Scholar] [CrossRef]
- Ashkar, A.A.; Yao, X.D.; Gill, N.; Sajic, D.; Patrick, A.J.; Rosenthal, K.L. Toll-like receptor (TLR)-3, but not TLR4, agonist protects against genital herpes infection in the absence of inflammation seen with CpG DNA. J. Infect. Dis. 2004, 190, 1841–1849. [Google Scholar] [CrossRef]
- Boivin, N.; Sergerie, Y.; Rivest, S.; Boivin, G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J. Infect. Dis. 2008, 198, 664–672. [Google Scholar] [CrossRef]
- Lai, Y.; Adhikarakunnathu, S., Bhardwaj; et al. LL37 and cationic peptides enhance TLR3 signaling by viral double-stranded RNAs. PLoS ONE 2011, 6, e26632. [Google Scholar]
- Lai, Y.; Yi, G.; Chen, A.; et al. Viral double-strand RNA-binding proteins can enhance innate immune signaling by toll-like Receptor 3. PLoS ONE 2011, 6, e25837. [Google Scholar]
- Funderburg, N.; Luciano, A.A.; Jiang, W.; Rodriguez, B.; Sieg, S.F.; Lederman, M.M. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS ONE 2008, 3, e1915. [Google Scholar] [CrossRef]
- Warger, T.; Osterloh, P.; Rechtsteiner, G.; et al. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood 2006, 108, 544–550. [Google Scholar] [CrossRef]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef]
- Gil-Torregrosa, B.C.; Lennon-Dumenil, A.M.; Kessler, B.; et al. Control of cross-presentation during dendritic cell maturation. Eur. J. Immunol. 2004, 34, 398–407. [Google Scholar] [CrossRef]
- Wilson, N.S.; Behrens, G.M.; Lundie, R.J.; et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 2006, 7, 165–172. [Google Scholar]
- Ashkar, A.A.; Mossman, K.L.; Coombes, B.K.; Gyles, C.L.; Mackenzie, R. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog 2008, 4, e1000233. [Google Scholar] [CrossRef]
- Equils, O.; Salehi, K.K.; Cornataeanu, R.; et al. Repeated lipopolysaccharide (LPS) exposure inhibits HIV replication in primary human macrophages. Microbes. Infect. 2006, 8, 2469–2476. [Google Scholar] [CrossRef]
- Equils, O.; Faure, E.; Thomas, L.; Bulut, Y.; Trushin, S.; Arditi, M. Bacterial lipopolysaccharide activates HIV long terminal repeat through Toll-like receptor 4. J. Immunol. 2001, 166, 2342–2347. [Google Scholar]
- Berg, R.S.; Aggerholm, A.; Bertelsen, L.S.; Ostergaard, L.; Paludan, S.R. Role of mitogen-activated protein kinases, nuclear factor-kappaB, and interferon regulatory factor 3 in Toll-like receptor 4-mediated activation of HIV long terminal repeat. APMIS 2009, 117, 124–132. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Wagner, T.L.; Ahonen, C.L.; Couture, A.M.; et al. Modulation of TH1 and TH2 cytokine production with the immune response modifiers, R-848 and imiquimod. Cell Immunol. 1999, 191, 10–19. [Google Scholar] [CrossRef]
- Lore, K.; Betts, M.R.; Brenchley, J.M.; et al. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J. Immunol. 2003, 171, 4320–4328. [Google Scholar]
- Prins, R.M.; Craft, N.; Bruhn, K.W.; et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J. Immunol. 2006, 176, 157–164. [Google Scholar]
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038. [Google Scholar] [CrossRef]
- Hart, O.M.; Athie-Morales, V.; O'connor, G.M.; Gardiner, C.M. TLR7/8-Mediated Activation of Human NK Cells Results in Accessory Cell-Dependent IFN-{gamma} Production. J. Immunol. 2005, 175, 1636–1642. [Google Scholar]
- Ablasser, A.; Poeck, H.; Anz, D.; et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J. Immunol. 2009, 182, 6824–6833. [Google Scholar] [CrossRef]
- Mark, K.E.; Corey, L.; Meng, T.C.; et al. Topical resiquimod 0.01% gel decreases herpes simplex virus type 2 genital shedding: A randomized, controlled trial. J. Infect. Dis. 2007, 195, 1324–1331. [Google Scholar]
- McCluskie, M.J.; Cartier, J.L.; Patrick, A.J.; et al. Treatment of intravaginal HSV-2 infection in mice: A comparison of CpG oligodeoxynucleotides and resiquimod (R-848). Antiviral Res. 2006, 69, 77–85. [Google Scholar] [CrossRef]
- Hammerbeck, D.M.; Burleson, G.R.; Schuller, C.J.; et al. Administration of a dual toll-like receptor 7 and toll-like receptor 8 agonist protects against influenza in rats. Antiviral Res. 2007, 73, 1–11. [Google Scholar] [CrossRef]
- Wille-Reece, U.; Flynn, B.J.; Lore, K.; et al. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. J. Exp. Med. 2006, 203, 1249–1258. [Google Scholar] [CrossRef]
- Wille-Reece, U.; Wu, C.Y.; Flynn, B.J.; Kedl, R.M.; Seder, R.A. Immunization with HIV-1 Gag protein conjugated to a TLR7/8 agonist results in the generation of HIV-1 Gag-specific Th1 and CD8+ T cell responses. J. Immunol. 2005, 174, 7676–7683. [Google Scholar]
- Velasquez, L.S.; Hjelm, B.E.; Arntzen, C.J.; Herbst-Kralovetz, M.M. An intranasally delivered Toll-like receptor 7 agonist elicits robust systemic and mucosal responses to Norwalk virus-like particles. Clin. Vaccine Immunol. 2010, 17, 1850–1858. [Google Scholar] [CrossRef]
- Weeratna, R.D.; Makinen, S.R.; McCluskie, M.J.; Davis, H.L. TLR agonists as vaccine adjuvants: comparison of CpG ODN and Resiquimod (R-848). Vaccine 2005, 23, 5263–5270. [Google Scholar]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef]
- Khairuddin, N.; Gantier, M.P.; Blake, S.J.; et al. siRNA-induced immunostimulation through TLR7 promotes antitumoral activity against HPV-driven tumors in vivo. Immunol. Cell Biol. 2011, 90, 187–196. [Google Scholar]
- Mureith, M.W.; Chang, J.J.; Lifson, J.D.; Ndung'u, T.; Altfeld, M. Exposure to HIV-1-encoded Toll-like receptor 8 ligands enhances monocyte response to microbial encoded Toll-like receptor 2/4 ligands. AIDS 2010, 24, 1841–1848. [Google Scholar] [CrossRef]
- Bukh, A.R.; Melchjorsen, J.; Offersen, R.; et al. Endotoxemia is associated with altered innate and adaptive immune responses in untreated HIV-1 infected individuals. PLoS ONE 2011, 6, e21275. [Google Scholar]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug. Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef]
- Kerkmann, M.; Rothenfusser, S.; Hornung, V.; et al. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J. Immunol. 2003, 170, 4465–4474. [Google Scholar]
- Becker, Y. CpG ODNs treatments of HIV-1 infected patients may cause the decline of transmission in high risk populations - a review, hypothesis and implications. Virus Genes 2005, 30, 251–266. [Google Scholar] [CrossRef]
- Gallichan, W.S.; Woolstencroft, R.N.; Guarasci, T.; McCluskie, M.J.; Davis, H.L.; Rosenthal, K.L. Intranasal immunization with CpG oligodeoxynucleotides as an adjuvant dramatically increases IgA and protection against herpes simplex virus-2 in the genital tract. J. Immunol. 2001, 166, 3451–3457. [Google Scholar]
- Sajic, D.; Ashkar, A.A.; Patrick, A.J.; et al. Parameters of CpG oligodeoxynucleotide-induced protection against intravaginal HSV-2 challenge. J. Med. Virol. 2003, 71, 561–568. [Google Scholar] [CrossRef]
- Harandi, A.M. The potential of immunostimulatory CpG DNA for inducing immunity against genital herpes: opportunities and challenges. J. Clin. Virol. 2004, 30, 207–210. [Google Scholar] [CrossRef]
- Equils, O.; Schito, M.L.; Karahashi, H.; et al. Toll-like receptor 2 (TLR2) and TLR9 signaling results in HIV-long terminal repeat trans-activation and HIV replication in HIV-1 transgenic mouse spleen cells: Implications of simultaneous activation of TLRs on HIV replication. J. Immunol. 2003, 170, 5159–5164. [Google Scholar]
- Jensen, K.M.; Melchjorsen, J.; Dagnaes-Hansen, F.; et al. Timing of toll-like receptor 9 agonist administration in pneumococcal vaccination impact both humoral and cellular immune responses as well as nasopharyngeal colonization in mice. Infect. Immun. 2012, 80, 1744–1752. [Google Scholar] [CrossRef]
- Pichlmair, A.; Diebold, S.S.; Gschmeissner, S.; et al. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J. Virol. 2007, 81, 539–547. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef]
- Coulombe, F.; Fiola, S.; Akira, S.; Cormier, Y.; Gosselin, J. Muramyl dipeptide induces NOD2-dependent Ly6C(high) monocyte recruitment to the lungs and protects against influenza virus infection. PLoS ONE 2012, 7, e36734. [Google Scholar]
- Shafique, M.; Wilschut, J.; de, H.A. Induction of mucosal and systemic immunity against respiratory syncytial virus by inactivated virus supplemented with TLR9 and NOD2 ligands. Vaccine 2012, 30, 597–606. [Google Scholar] [CrossRef]
- Zaks, K.; Jordan, M.; Guth, A.; et al. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J. Immunol. 2006, 176, 7335–7345. [Google Scholar]
- Bernstein, D.I.; Cardin, R.D.; Bravo, F.J.; et al. Potent adjuvant activity of cationic liposome-DNA complexes for genital herpes vaccines. Clin. Vaccine Immunol. 2009, 16, 699–705. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Farley, N.; Bravo, F.J.; et al. The adjuvant CLDC increases protection of a herpes simplex type 2 glycoprotein D vaccine in guinea pigs. Vaccine 2010, 28, 3748–3753. [Google Scholar] [CrossRef]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.G.; et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog 2009, 5, e1000480. [Google Scholar] [CrossRef]
- Lladser, A.; Mougiakakos, D.; Tufvesson, H.; et al. DAI (DLM-1/ZBP1) as a genetic adjuvant for DNA vaccines that promotes effective antitumor CTL immunity. Mol. Ther. 2011, 19, 594–601. [Google Scholar] [CrossRef]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef]
- Huang, L.; Lemos, H.P.; Li, L.; et al. Engineering DNA nanoparticles as immunomodulatory teagents that activate regulatory T cells. J. Immunol. 2012, 188, 4913–4920. [Google Scholar] [CrossRef]
- Equils, O.; Shapiro, A.; Madak, Z.; Liu, C.; Lu, D. Human immunodeficiency virus type 1 protease inhibitors block toll-like receptor 2 (TLR2)- and TLR4-Induced NF-kappaB activation. Antimicrob. Agents. Chemother. 2004, 48, 3905–3911. [Google Scholar] [CrossRef]
- Wallet, M.A.; Reist, C.M.; Williams, J.C.; et al. The HIV-1 protease inhibitor nelfinavir activates PP2 and inhibits MAPK signaling in macrophages: A pathway to reduce inflammation. J. Leukoc. Biol. 2012, 92, 795–805. [Google Scholar] [CrossRef]
- Danaher, R.J.; Kaetzel, C.S.; Greenberg, R.N.; Wang, C.; Bruno, M.E.; Miller, C.S. HIV protease inhibitors alter innate immune response signaling to double-stranded RNA in oral epithelial cells: Implications for immune reconstitution inflammatory syndrome? AIDS 2010, 24, 2587–2590. [Google Scholar]
- Pajonk, F.; Himmelsbach, J.; Riess, K.; Sommer, A.; McBride, W.H. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res. 2002, 62, 5230–5235. [Google Scholar]
- Kurata, S. Potential of azidothymidine to activate the HIV-1 promoter. J. Biol. Chem. 1994, 269, 24553–24556. [Google Scholar]
- Melchjorsen, J.; Risør, M.W.; Sogaard, O.S.; et al. Tenofovir selectively regulates production of inflammatory cytokines and shifts the IL-12 / IL-10 balance in human primary cells. J. Acquir. Immune. Defic. Syndr. 2011, 57, 265–275. [Google Scholar] [CrossRef]
- Kurokawa, M.; Ghosh, S.K.; Ramos, J.C.; et al. Azidothymidine inhibits NF-kappaB and induces Epstein-Barr virus gene expression in Burkitt lymphoma. Blood 2005, 106, 235–240. [Google Scholar] [CrossRef]
- Martin, A.M.; Almeida, C.A.; Cameron, P.; et al. Immune responses to abacavir in antigen-presenting cells from hypersensitive patients. AIDS 2007, 21, 1233–1244. [Google Scholar] [CrossRef]
- Van Rompay, K.K.; Marthas, M.L.; Bischofberger, N. Tenofovir primes rhesus macaque cells in vitro for enhanced interleukin-12 secretion. Antiviral Res. 2004, 63, 133–138. [Google Scholar] [CrossRef]
- Zidek, Z.; Frankova, D.; Holy, A. Activation by 9-(R)-[2-(phosphonomethoxy)propyl]adenine of chemokine (RANTES, macrophage inflammatory protein 1alpha) and cytokine (tumor necrosis factor alpha, interleukin-10 [IL-10], IL-1beta) production. Antimicrob. Agents. Chemother. 2001, 45, 3381–3386. [Google Scholar] [CrossRef]
- Zidek, Z.; Kmonickova, E.; Holy, A. Secretion of antiretroviral chemokines by human cells cultured with acyclic nucleoside phosphonates. Eur. J. Pharmacol. 2007, 574, 77–84. [Google Scholar] [CrossRef]
- Abdool, K.Q.; Abdool Karim, S.S.; Frohlich, J.A.; et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010, 329, 1168–1174. [Google Scholar] [CrossRef]
- FHI and the Centre for the AIDS Programme of Research in South Africa; Factsheet: CAPRISA 004 Trial and the impact of tenofovir gel on herpes simplex virus type-2 infections; Research Triangle Park: NC, USA, 2010.
- Mesquita, P.M.; Rastogi, R.; Segarra, T.J.; et al. Intravaginal ring delivery of tenofovir disoproxil fumarate for prevention of HIV and herpes simplex virus infection. J. Antimicrob. Chemother. 2012, 67, 1730–1738. [Google Scholar] [CrossRef]
- Andrei, G.; Lisco, A.; Vanpouille, C.; et al. Topical tenofovir, a microbicide effective against HIV, inhibits herpes simplex virus-2 replication. Cell Host Microbe 2011, 10, 379–389. [Google Scholar] [CrossRef]
- Vibholm, L.; Reinert, L.S.; Sogaard, O.S.; et al. Antiviral and immunological effects of tenofovir microbicide in vaginal herpes simplex virus 2 infection. AIDS Res. Hum. Retroviruses 2012, 28, 1404–1411. [Google Scholar] [CrossRef]
- Kikuchi, T.; Hagiwara, K.; Honda, Y.; et al. Clarithromycin suppresses lipopolysaccharide-induced interleukin-8 production by human monocytes through AP-1 and NF-kappa B transcription factors. J. Antimicrob. Chemother. 2002, 49, 745–755. [Google Scholar] [CrossRef]
- Khair, O.A.; Devalia, J.L.; Abdelaziz, M.M.; Sapsford, R.J.; Davies, R.J. Effect of erythromycin on Haemophilus influenzae endotoxin-induced release of IL-6, IL-8 and sICAM-1 by cultured human bronchial epithelial cells. Eur. Respir. J. 1995, 8, 1451–1457. [Google Scholar]
- Schultz, M.J.; Speelman, P.; Van Der Poll, T. Erythromycin inhibits Pseudomonas aeruginosa-induced tumour necrosis factor-alpha production in human whole blood. J. Antimicrob. Chemother. 2001, 48, 275–278. [Google Scholar] [CrossRef]
- Li, D.Q.; Zhou, N.; Zhang, L.; Ma, P.; Pflugfelder, S.C. Suppressive effects of azithromycin on zymosan-induced production of proinflammatory mediators by human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5623–5629. [Google Scholar] [CrossRef]
- Murphy, D.M.; Forrest, I.A.; Corris, P.A.; et al. Azithromycin attenuates effects of lipopolysaccharide on lung allograft bronchial epithelial cells. J. Heart Lung Transplant. 2008, 27, 1210–1216. [Google Scholar] [CrossRef]
- Morikawa, K.; Zhang, J.; Nonaka, M.; Morikawa, S. Modulatory effect of macrolide antibiotics on the Th1- and Th2-type cytokine production. Int. J. Antimicrob. Agents. 2002, 19, 53–59. [Google Scholar] [CrossRef]
- Murphy, B.S.; Sundareshan, V.; Cory, T.J.; Hayes, D., Jr.; Anstead, M.I.; Feola, D.J. Azithromycin alters macrophage phenotype. J. Antimicrob. Chemother. 2008, 61, 554–560. [Google Scholar] [CrossRef]
- Darisipudi, M.N.; Allam, R.; Rupanagudi, K.V.; Anders, H.J. Polyene Macrolide Antifungal Drugs Trigger Interleukin-1beta Secretion by Activating the NLRP3 Inflammasome. PLoS ONE 2011, 6, e19588. [Google Scholar]
- Razonable, R.R.; Henault, M.; Watson, H.L.; Paya, C.V. Nystatin induces secretion of interleukin (IL)-1beta, IL-8, and tumor necrosis factor alpha by a toll-like receptor-dependent mechanism. Antimicrob. Agents. Chemother. 2005, 49, 3546–3549. [Google Scholar] [CrossRef]
- Razonable, R.R.; Henault, M.; Lee, L.N.; et al. Secretion of proinflammatory cytokines and chemokines during amphotericin B exposure is mediated by coactivation of toll-like receptors 1 and 2. Antimicrob. Agents. Chemother. 2005, 49, 1617–1621. [Google Scholar] [CrossRef]
- Rizzo, A.; Paolillo, R.; Guida, L.; Annunziata, M.; Bevilacqua, N.; Tufano, M.A. Effect of metronidazole and modulation of cytokine production on human periodontal ligament cells. Int. Immunopharmacol. 2010, 10, 744–750. [Google Scholar] [CrossRef]
- Hirata, N.; Hiramatsu, K.; Kishi, K.; Yamasaki, T.; Ichimiya, T.; Nasu, M. Pretreatment of mice with clindamycin improves survival of endotoxic shock by modulating the release of inflammatory cytokines. Antimicrob. Agents. Chemother. 2001, 45, 2638–2642. [Google Scholar] [CrossRef]
- Nakano, T.; Hiramatsu, K.; Kishi, K.; Hirata, N.; Kadota, J.; Nasu, M. Clindamycin modulates inflammatory-cytokine induction in lipopolysaccharide-stimulated mouse peritoneal macrophages. Antimicrob. Agents. Chemother. 2003, 47, 363–367. [Google Scholar]
- Araujo, F.G.; Slifer, T.L.; Remington, J.S. Inhibition of secretion of interleukin-1alpha and tumor necrosis factor alpha by the ketolide antibiotic telithromycin. Antimicrob. Agents. Chemother. 2002, 46, 3327–3330. [Google Scholar] [CrossRef]
- Sugiura, Y.; Hiramatsu, K.; Hamauzu, R.; et al. Mitogen-activated protein kinases-dependent induction of hepatocyte growth factor production in human dermal fibroblasts by the antibiotic polymyxin B. Cytokine 2012, 60, 205–211. [Google Scholar]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Melchjorsen, J. Learning from the Messengers: Innate Sensing of Viruses and Cytokine Regulation of Immunity — Clues for Treatments and Vaccines. Viruses 2013, 5, 470-527. https://doi.org/10.3390/v5020470
Melchjorsen J. Learning from the Messengers: Innate Sensing of Viruses and Cytokine Regulation of Immunity — Clues for Treatments and Vaccines. Viruses. 2013; 5(2):470-527. https://doi.org/10.3390/v5020470
Chicago/Turabian StyleMelchjorsen, Jesper. 2013. "Learning from the Messengers: Innate Sensing of Viruses and Cytokine Regulation of Immunity — Clues for Treatments and Vaccines" Viruses 5, no. 2: 470-527. https://doi.org/10.3390/v5020470
APA StyleMelchjorsen, J. (2013). Learning from the Messengers: Innate Sensing of Viruses and Cytokine Regulation of Immunity — Clues for Treatments and Vaccines. Viruses, 5(2), 470-527. https://doi.org/10.3390/v5020470