Early Events in Foamy Virus—Host Interaction and Intracellular Trafficking


{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Early Events in Virus-Host Interaction: Attachment and Entry
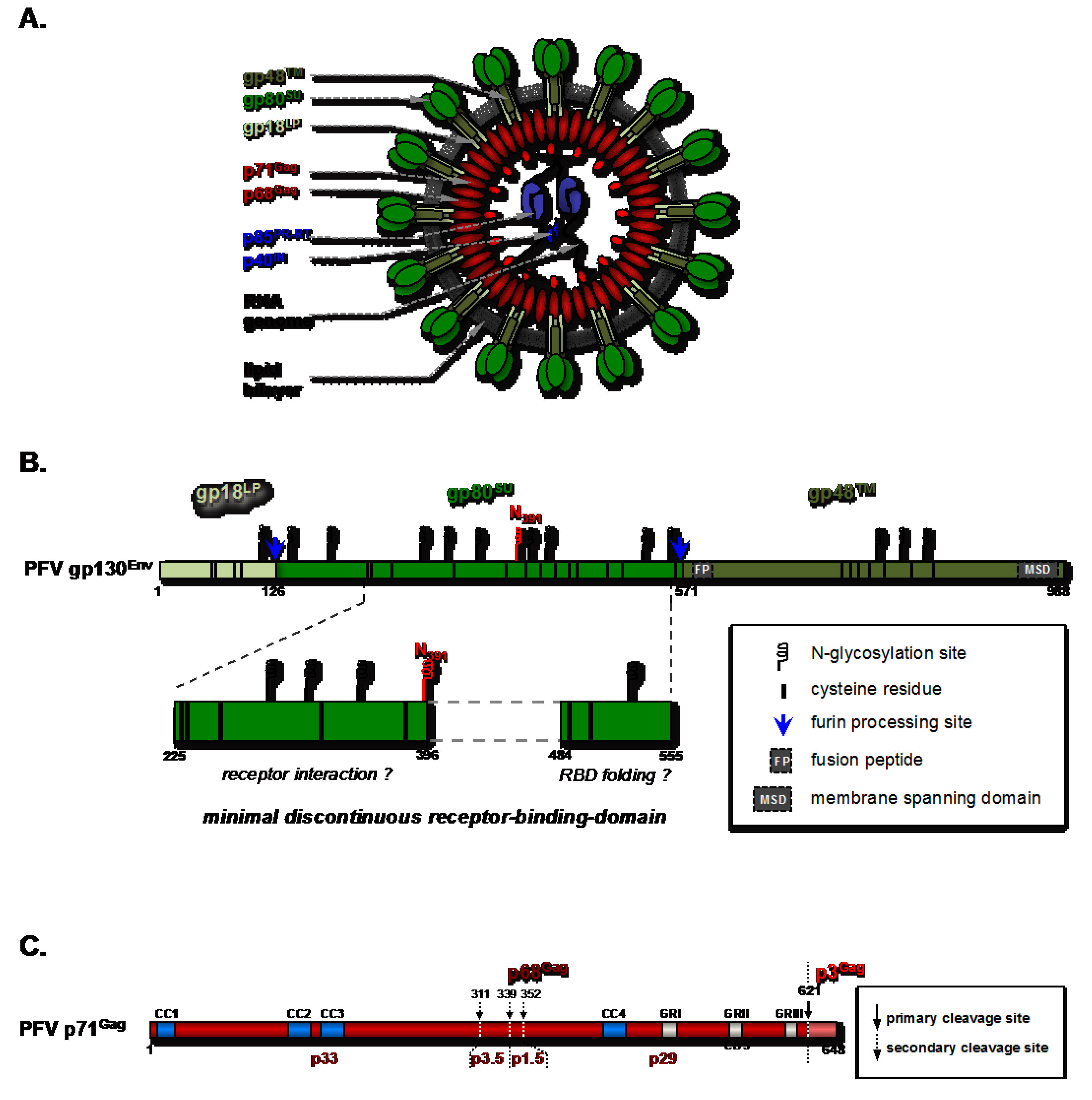
2.1. Molecular Determinants of the Foamy Viral Glycoprotein for Attachment and Entry
2.1.1. FV-Env Mediated Superinfection Resistance
2.1.2. The FV Receptor-Binding-Domain and Its Functional Dependence on Post-Translational Modifications
2.2. Cellular Determinants for FV Attachment and Uptake

2.3. FV Env-Dependent Fusogenic Release of Viral Capsids
3. Post Fusion Events in FV Infection: Intracellular Trafficking, Disassembly and Formation of the Preintregration Complex
3.1. FV Capsid Trafficking towards the Microtubule-Organizing Center
3.2. Cell Cycle Dependence and Essential Components for Genome Integration
3.3. Capsid Disassembly—A Concerted Process Involving Cellular and Viral Enzymes?
4. Innate Sensing and Cellular Restriction Factors of FVs
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Hill, C.L.; Bieniasz, P.D.; McClure, M.O. Properties of human foamy virus relevant to its development as a vector for gene therapy. J. Gen. Virol. 1999, 80, 2003–2009. [Google Scholar]
- Mergia, A.; Leung, N.J.; Blackwell, J. Cell tropism of the simian foamy virus type 1 (SFV-1). J. Med. Primatol. 1996, 25, 2–7. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef]
- Wilk, T.; de Haas, F.; Wagner, A.; Rutten, T.; Fuller, S.; Flügel, R.M.; Löchelt, M. The intact retroviral Env glycoprotein of human foamy virus is a trimer. J. Virol. 2000, 74, 2885–2887. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Persons, D.A. Cell membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol. Ther. 2012, 20, 1158–1166. [Google Scholar] [CrossRef]
- Plochmann, K.; Horn, A.; Gschmack, E.; Armbruster, N.; Krieg, J.; Wiktorowicz, T.; Weber, C.; Stirnnagel, K.; Lindemann, D.; Rethwilm, A.; et al. Heparan sulfate is an attachment factor for foamy virus entry. J. Virol. 2012, 86, 10028–10035. [Google Scholar] [CrossRef]
- Stirnnagel, K.; Lüftenegger, D.; Stange, A.; Swiersy, A.; Müllers, E.; Reh, J.; Stanke, N.; Grosse, A.; Chiantia, S.; Keller, H.; et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 2010, 7, e45. [Google Scholar]
- Lee, E.G.; Stenbak, C.R.; Linial, M.L. Foamy virus assembly with emphasis on pol encapsidation. Viruses 2013, 5, 886–900. [Google Scholar] [CrossRef]
- Hütter, S.; Zurnic, I.; Lindemann, D. Foamy virus budding and release. Viruses 2013, 5. submitted. [Google Scholar]
- Lehmann-Che, J.; Renault, N.; Giron, M.L.; Roingeard, P.; Clave, E.; Tobaly-Tapiero, J.; Bittoun, P.; Toubert, A.; de The, H.; Saib, A. Centrosomal latency of incoming foamy viruses in resting cells. PLoS Pathog. 2007, 3, e74. [Google Scholar] [CrossRef]
- Petit, C.; Giron, M.L.; Tobaly-Tapiero, J.; Bittoun, P.; Real, E.; Jacob, Y.; Tordo, N.; de The, H.; Saib, A. Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J. Cell Sci. 2003, 116, 3433–3442. [Google Scholar] [CrossRef]
- Saib, A.; Puvion Dutilleul, F.; Schmid, M.; Peries, J.; de The, H. Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J. Virol. 1997, 71, 1155–1161. [Google Scholar]
- Stirnnagel, K.; Schupp, D.; Dupont, A.; Kudryavtsev, V.; Reh, J.; Mullers, E.; Lamb, D.C.; Lindemann, D. Differential pH-dependent cellular uptake pathways among foamy viruses elucidated using dual-colored fluorescent particles. Retrovirology 2012, 9, e71. [Google Scholar]
- Hütter, S.; Müllers, E.; Stanke, N.; Reh, J.; Lindemann, D. Prototype Foamy Virus protease activity is essential for intra-particle reverse transcription initiation but not absolutely required for uncoating upon host cell entry. J. Virol. 2013, in press. [Google Scholar]
- Lehmann-Che, J.; Giron, M.L.; Delelis, O.; Lochelt, M.; Bittoun, P.; Tobaly-Tapiero, J.; de The, H.; Saib, A. Protease-dependent uncoating of a complex retrovirus. J. Virol. 2005, 79, 9244–9253. [Google Scholar] [CrossRef]
- Bieniasz, P.D.; Weiss, R.A.; McClure, M.O. Cell cycle dependence of foamy retrovirus infection. J. Virol. 1995, 69, 7295–7299. [Google Scholar]
- Lo, Y.T.; Tian, T.; Nadeau, P.E.; Park, J.; Mergia, A. The foamy virus genome remains unintegrated in the nuclei of G1/S phase-arrested cells, and integrase is critical for preintegration complex transport into the nucleus. J. Virol. 2010, 84, 2832–2842. [Google Scholar] [CrossRef]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef]
- Nethe, M.; Berkhout, B.; van der Kuyl, A.C. Retroviral superinfection resistance. Retrovirology 2005, 2, e52. [Google Scholar]
- Sommerfelt, M.A.; Weiss, R.A. Receptor interference groups of 20 retroviruses plating on human cells. Virology 1990, 176, 58–69. [Google Scholar] [CrossRef]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Berg, A.; Pietschmann, T.; Rethwilm, A.; Lindemann, D. Determinants of foamy virus envelope glycoprotein mediated resistance to superinfection. Virology 2003, 314, 243–252. [Google Scholar] [CrossRef]
- Herchenröder, O.; Moosmayer, D.; Bock, M.; Pietschmann, T.; Rethwilm, A.; Bieniasz, P.D.; McClure, M.O.; Weis, R.; Schneider, J. Specific binding of recombinant foamy virus envelope protein to host cells correlates with susceptibility to infection. Virology 1999, 255, 228–236. [Google Scholar] [CrossRef]
- Duda, A.; Luftenegger, D.; Pietschmann, T.; Lindemann, D. Characterization of the prototype foamy virus envelope glycoprotein receptor-binding domain. J. Virol. 2006, 80, 8158–8167. [Google Scholar] [CrossRef]
- Hunter, E. Viral Entry and Receptors. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1997. [Google Scholar]
- Battini, J.L.; Danos, O.; Heard, J.M. Receptor-binding domain of murine leukemia virus envelope glycoproteins. J. Virol. 1995, 69, 713–719. [Google Scholar]
- Davey, R.A.; Hamson, C.A.; Healey, J.J.; Cunningham, J.M. In vitro binding of purified murine ecotropic retrovirus envelope surface protein to its receptor, MCAT-1. J. Virol. 1997, 71, 8096–8102. [Google Scholar]
- Heard, J.M.; Danos, O. An amino-terminal fragment of the Friend murine leukemia virus envelope glycoprotein binds the ecotropic receptor. J. Virol. 1991, 65, 4026–4032. [Google Scholar]
- Kowalski, M.; Potz, J.; Basiripour, L.; Dorfman, T.; Goh, W.C.; Terwilliger, E.; Dayton, A.; Rosen, C.; Haseltine, W.; Sodroski, J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 1987, 237, 1351–1355. [Google Scholar]
- Nygren, A.; Bergman, T.; Matthews, T.; Jornvall, H.; Wigzell, H. 95- and 25-kDa fragments of the human immunodeficiency virus envelope glycoprotein gp120 bind to the CD4 receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 6543–6546. [Google Scholar] [CrossRef]
- Sun, Y.; Wen, D.D.; Liu, Q.M.; Yi, X.F.; Wang, T.T.; Wei, L.L.; Li, Z.; Liu, W.H.; He, X.H. Comparative analysis of the envelope glycoproteins of foamy viruses. Acta Virol. 2012, 56, 283–291. [Google Scholar] [CrossRef]
- Lüftenegger, D.; Picard-Maureau, M.; Stanke, N.; Rethwilm, A.; Lindemann, D. Analysis and function of prototype foamy virus envelope N glycosylation. J. Virol. 2005, 79, 7664–7672. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Krey, T.; d'Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef] [Green Version]
- Wahid, A.; Helle, F.; Descamps, V.; Duverlie, G.; Penin, F.; Dubuisson, J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J. Virol. 2013, 87, 1605–1617. [Google Scholar] [CrossRef]
- McCaffrey, K.; Boo, I.; Tewierek, K.; Edmunds, M.L.; Poumbourios, P.; Drummer, H.E. Role of conserved cysteine residues in hepatitis C virus glycoprotein e2 folding and function. J. Virol. 2012, 86, 3961–3974. [Google Scholar] [CrossRef]
- Mergia, A.; Shaw, K.E.; Lackner, J.E.; Luciw, P.A. Relationship of the env genes and the endonuclease domain of the pol genes of simian foamy virus type 1 and human foamy virus. J. Virol. 1990, 64, 406–410. [Google Scholar]
- Crooks, G.M.; Kohn, D.B. Growth factors increase amphotropic retrovirus binding to human CD34+ bone marrow progenitor cells. Blood 1993, 82, 3290–3297. [Google Scholar]
- Liu, J.; Thorp, S.C. Cell surface heparan sulfate and its roles in assisting viral infections. Med. Res. Rev. 2002, 22, 1–25. [Google Scholar] [CrossRef]
- Peisajovich, S.G.; Shai, Y. High similarity between reverse-oriented sequences from HIV and foamy virus envelope glycoproteins. AIDS Res. Hum. Retrovir. 2002, 18, 309–312. [Google Scholar] [CrossRef]
- Wang, G.; Mulligan, M.J. Comparative sequence analysis and predictions for the envelope glycoproteins of foamy viruses. J. Gen. Virol. 1999, 80, 245–254. [Google Scholar]
- Pietschmann, T.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. An evolutionarily conserved positively charged amino acid in the putative membrane-spanning domain of the foamy virus envelope protein controls fusion activity. J. Virol. 2000, 74, 4474–4482. [Google Scholar] [CrossRef]
- Bansal, A.; Shaw, K.L.; Edwards, B.H.; Goepfert, P.A.; Mulligan, M.J. Characterization of the R572T point mutant of a putative cleavage site in human foamy virus Env. J. Virol. 2000, 74, 2949–2954. [Google Scholar] [CrossRef]
- Duda, A.; Stange, A.; Luftenegger, D.; Stanke, N.; Westphal, D.; Pietschmann, T.; Eastman, S.W.; Linial, M.L.; Rethwilm, A.; Lindemann, D. Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity. J. Virol. 2004, 78, 13865–13870. [Google Scholar] [CrossRef]
- Geiselhart, V.; Bastone, P.; Kempf, T.; Schnolzer, M.; Löchelt, M. Furin-mediated cleavage of the feline foamy virus Env leader protein. J. Virol. 2004, 78, 13573–13581. [Google Scholar] [CrossRef]
- Picard-Maureau, M.; Jarmy, G.; Berg, A.; Rethwilm, A.; Lindemann, D. Foamy virus envelope glycoprotein-mediated entry involves a pH-Dependent fusion process. J. Virol. 2003, 77, 4722–4730. [Google Scholar] [CrossRef]
- Radtke, K.; Dohner, K.; Sodeik, B. Viral interactions with the cytoskeleton: A hitchhiker’s guide to the cell. Cell Microbiol. 2006, 8, 387–400. [Google Scholar] [CrossRef]
- Fischer, N.; Heinkelein, M.; Lindemann, D.; Enssle, J.; Baum, C.; Werder, E.; Zentgraf, H.; Müller, J.G.; Rethwilm, A. Foamy virus particle formation. J. Virol. 1998, 72, 1610–1615. [Google Scholar]
- Schliephake, A.W.; Rethwilm, A. Nuclear localization of foamy virus Gag precursor protein. J. Virol. 1994, 68, 4946–4954. [Google Scholar]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef]
- Schliwa, M.; Woehlke, G. Molecular motors. Nature 2003, 422, 759–765. [Google Scholar] [CrossRef]
- Giron, M.L.; Colas, S.; Wybier, J.; Rozain, F.; Emanoil Ravier, R. Expression and maturation of human foamy virus Gag precursor polypeptides. J. Virol. 1997, 71, 1635–1639. [Google Scholar]
- Stolp, B.; Fackler, O.T. How HIV takes advantage of the cytoskeleton in entry and replication. Viruses 2011, 3, 293–311. [Google Scholar] [CrossRef]
- Imrich, H.; Heinkelein, M.; Herchenroder, O.; Rethwilm, A. Primate foamy virus Pol proteins are imported into the nucleus. J. Gen. Virol. 2000, 81, 2941–2947. [Google Scholar]
- Hyun, U.; Lee, D.H.; Shin, C.G. Minimal size of prototype foamy virus integrase for nuclear localization. Acta Virol. 2011, 55, 169–174. [Google Scholar] [CrossRef]
- An, D.G.; Hyun, U.; Shin, C.G. Characterization of nuclear localization signals of the prototype foamy virus integrase. J. Gen. Virol. 2008, 89, 1680–1684. [Google Scholar]
- Tobaly-Tapiero, J.; Bittoun, P.; Lehmann-Che, J.; Delelis, O.; Giron, M.L.; de The, H.; Saib, A. Chromatin tethering of incoming foamy virus by the structural Gag protein. Traffic 2008, 9, 1717–1727. [Google Scholar] [CrossRef]
- Yu, S.F.; Edelmann, K.; Strong, R.K.; Moebes, A.; Rethwilm, A.; Linial, M.L. The carboxyl terminus of the human foamy virus Gag protein contains separable nucleic acid binding and nuclear transport domains. J. Virol. 1996, 70, 8255–8262. [Google Scholar]
- Mullers, E.; Stirnnagel, K.; Kaulfuss, S.; Lindemann, D. Prototype Foamy virus gag nuclear localization: A novel pathway among retroviruses. J. Virol. 2011, 85, 9276–9285. [Google Scholar] [CrossRef]
- Stenbak, C.R.; Linial, M.L. Role of the C terminus of foamy virus Gag in RNA packaging and Pol expression. J. Virol. 2004, 78, 9423–9430. [Google Scholar] [CrossRef]
- Müllers, E.; Uhlig, T.; Stirnnagel, K.; Fiebig, U.; Zentgraf, H.; Lindemann, D. Novel functions of prototype foamy virus Gag glycine- arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 2011, 85, 1452–1463. [Google Scholar] [CrossRef]
- Fassati, A. Multiple roles of the capsid protein in the early steps of HIV-1 infection. Virus Res. 2012, 170, 15–24. [Google Scholar] [CrossRef]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, e96. [Google Scholar] [CrossRef]
- Yu, S.F.; Baldwin, D.N.; Gwynn, S.R.; Yendapalli, S.; Linial, M.L. Human foamy virus replication: A pathway distinct from that of retroviruses and hepadnaviruses. Science 1996, 271, 1579–1582. [Google Scholar]
- Zamborlini, A.; Renault, N.; Saib, A.; Delelis, O. Early reverse transcription is essential for productive foamy virus infection. PLoS One 2010, 5, e11023. [Google Scholar] [CrossRef]
- Delelis, O.; Saib, A.; Sonigo, P. Biphasic DNA synthesis in spumaviruses. J. Virol. 2003, 77, 8141–8146. [Google Scholar] [CrossRef]
- Pfrepper, K.I.; Löchelt, M.; Rackwitz, H.R.; Schnolzer, M.; Heid, H.; Flügel, R.M. Molecular characterization of proteolytic processing of the Gag proteins of human spumavirus. J. Virol. 1999, 73, 7907–7911. [Google Scholar]
- Linial, M. Why aren’t foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Murray, S.M.; Picker, L.J.; Axthelm, M.K.; Hudkins, K.; Alpers, C.E.; Linial, M.L. Replication in a superficial epithelial cell niche explains the lack of pathogenicity of primate foamy virus infections. J. Virol. 2008, 82, 5981–5985. [Google Scholar] [CrossRef]
- Iwasaki, A. Innate immune recognition of HIV-1. Immunity 2012, 37, 389–398. [Google Scholar] [CrossRef]
- Ploquin, M.J.; Jacquelin, B.; Jochems, S.P.; Barre-Sinoussi, F.; Muller-Trutwin, M.C. Innate immunity in the control of HIV/AIDS: Recent advances and open questions. AIDS 2012, 26, 1269–1279. [Google Scholar] [CrossRef]
- Colas, S.; Bourge, J.F.; Wybier, J.; Chelbi Alix, M.K.; Paul, P.; Emanoil Ravier, R. Human foamy virus infection activates class I major histocompatibility complex antigen expression. J. Gen. Virol. 1995, 76, 661–667. [Google Scholar] [CrossRef]
- Rhodes-Feuillette, A.; Saal, F.; Lasneret, J.; Santillana-Hayat, M.; Peries, J. Studies on in vitro interferon induction capacity and interferon sensitivity of simian foamy viruses. Arch. Virol. 1987, 97, 77–84. [Google Scholar] [CrossRef]
- Sabile, A.; Rhodes Feuillette, A.; Jaoui, F.Z.; Tobaly Tapiero, J.; Giron, M.L.; Lasneret, J.; Peries, J.; Canivet, M. In vitro studies on interferon-inducing capacity and sensitivity to IFN of human foamy virus. Res. Virol. 1996, 147, 29–37. [Google Scholar] [CrossRef]
- Rua, R.; Lepelley, A.; Gessain, A.; Schwartz, O. Innate sensing of foamy viruses by human hematopoietic cells. J. Virol. 2012, 86, 909–918. [Google Scholar] [CrossRef]
- Meiering, C.D.; Linial, M.L. The promyelocytic leukemia protein does not mediate foamy virus latencyin vitro. J. Virol. 2003, 77, 2207–2213. [Google Scholar] [CrossRef]
- Regad, T.; Saib, A.; Lallemand-Breitenbach, V.; Pandolfi, P.P.; de The, H.; Chelbi-Alix, M.K. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 2001, 20, 3495–3505. [Google Scholar] [CrossRef]
- Matthes, D.; Wiktorowicz, T.; Zahn, J.; Bodem, J.; Stanke, N.; Lindemann, D.; Rethwilm, A. Basic residues in the foamy virus gag protein. J. Virol. 2011, 85, 3986–3995. [Google Scholar] [CrossRef]
- Xu, F.; Tan, J.; Liu, R.; Xu, D.; Li, Y.; Geng, Y.; Liang, C.; Qiao, W. Tetherin inhibits prototypic foamy virus release. Virol. J. 2011, 8, e198. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Delebecque, F.; Suspene, R.; Calattini, S.; Casartelli, N.; Saib, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Pacheco, B.; Finzi, A.; McGee-Estrada, K.; Sodroski, J. Species-Specific inhibition of foamy viruses from South American monkeys by New World Monkey TRIM5{alpha} proteins. J. Virol. 2010, 84, 4095–4099. [Google Scholar] [CrossRef]
- Yap, M.W.; Lindemann, D.; Stanke, N.; Reh, J.; Westphal, D.; Hanenberg, H.; Ohkura, S.; Stoye, J.P. Restriction of foamy viruses by primate Trim5alpha. J. Virol. 2008, 82, 5429–5439. [Google Scholar] [CrossRef]
- De Silva, S.; Wu, L. TRIM5 acts as more than a retroviral restriction factor. Viruses 2011, 13, 1204–1209. [Google Scholar] [CrossRef]
- Grutter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berka, U.; Hamann, M.V.; Lindemann, D. Early Events in Foamy Virus—Host Interaction and Intracellular Trafficking. Viruses 2013, 5, 1055-1074. https://doi.org/10.3390/v5041055
Berka U, Hamann MV, Lindemann D. Early Events in Foamy Virus—Host Interaction and Intracellular Trafficking. Viruses. 2013; 5(4):1055-1074. https://doi.org/10.3390/v5041055
Chicago/Turabian StyleBerka, Ursula, Martin Volker Hamann, and Dirk Lindemann. 2013. "Early Events in Foamy Virus—Host Interaction and Intracellular Trafficking" Viruses 5, no. 4: 1055-1074. https://doi.org/10.3390/v5041055
APA StyleBerka, U., Hamann, M. V., & Lindemann, D. (2013). Early Events in Foamy Virus—Host Interaction and Intracellular Trafficking. Viruses, 5(4), 1055-1074. https://doi.org/10.3390/v5041055