Architectural Insight into Inovirus-Associated Vectors (IAVs) and Development of IAV-Based Vaccines Inducing Humoral and Cellular Responses: Implications in HIV-1 Vaccines


Abstract
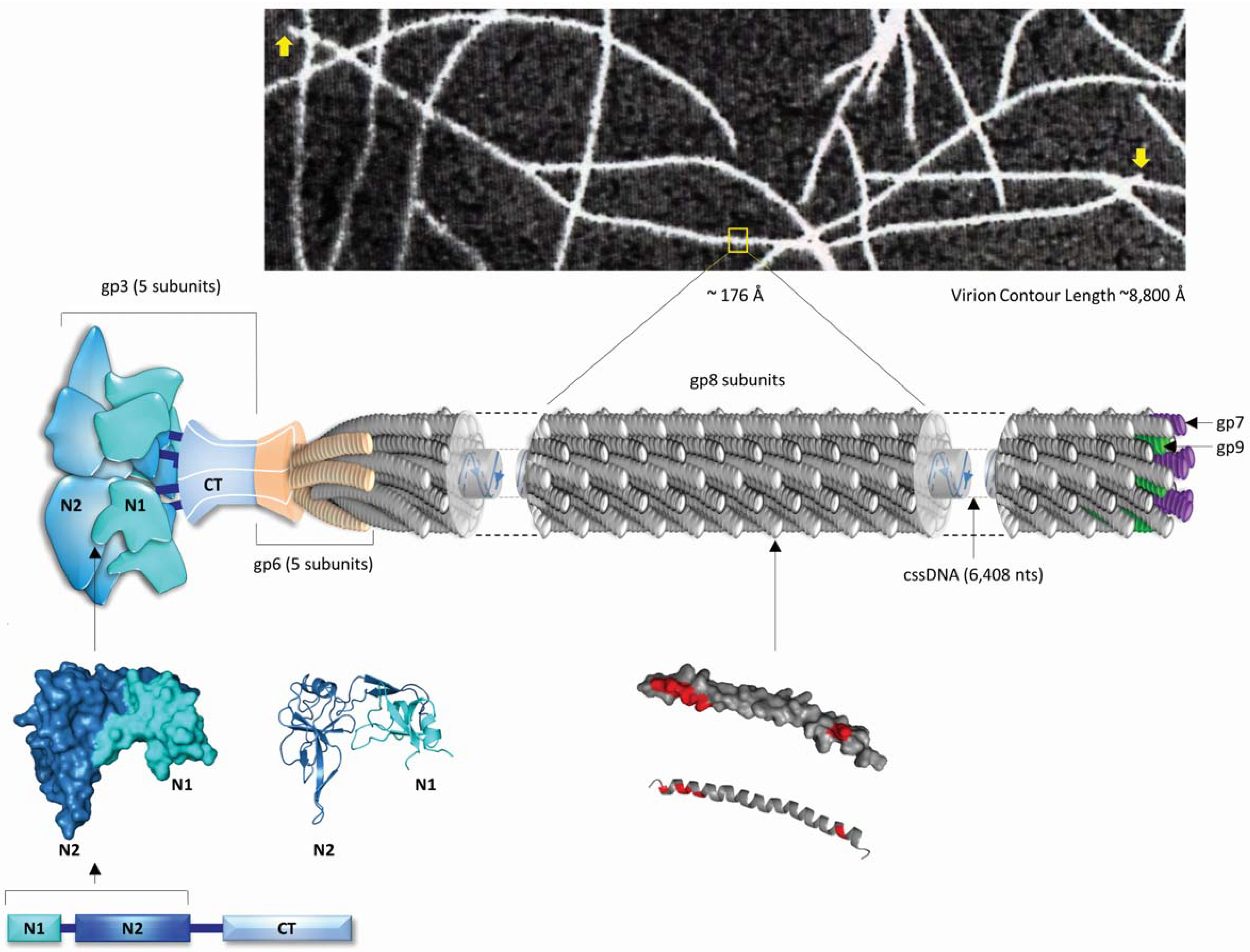
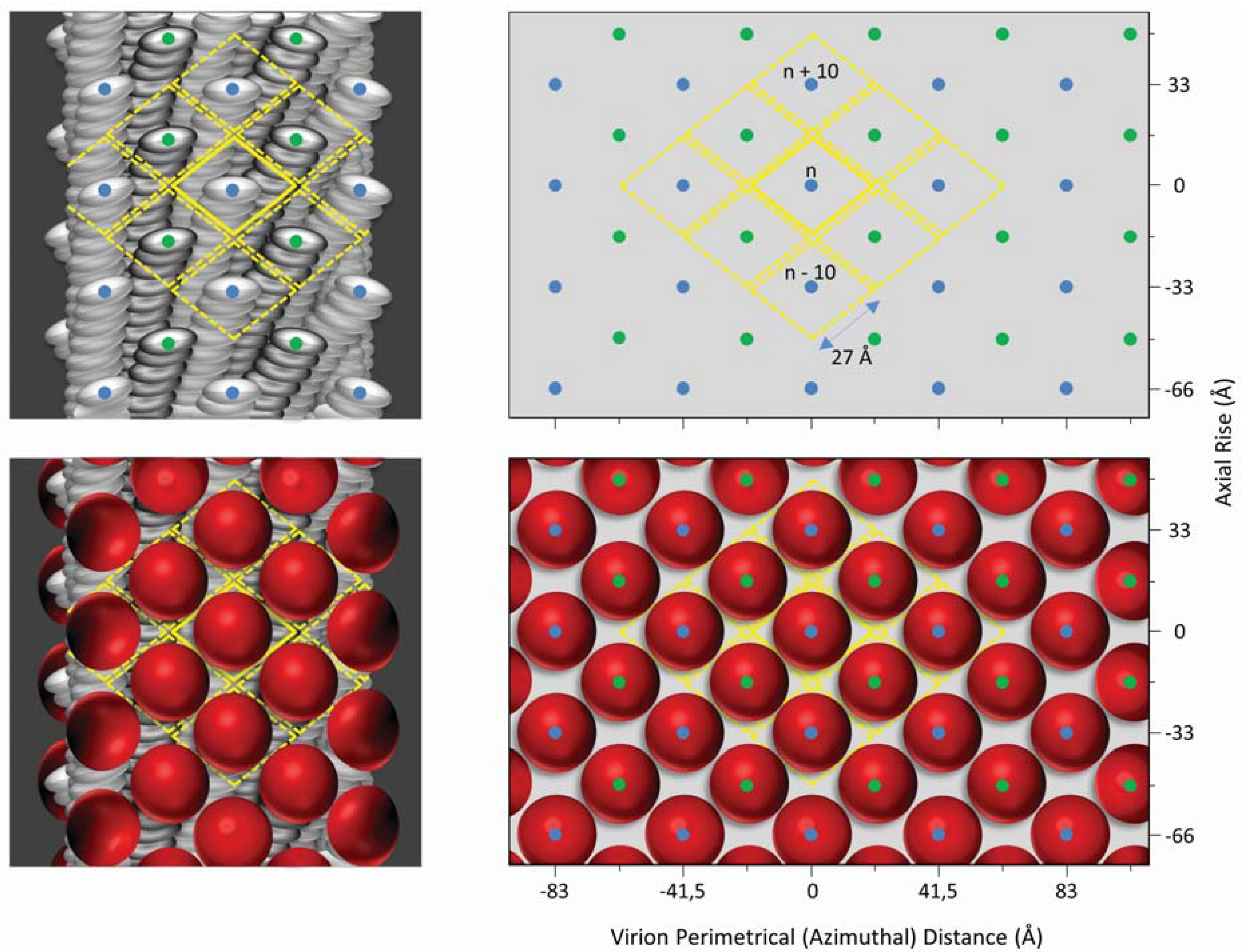
:1. Structural and Biological Insights Into Filamentous Viruses

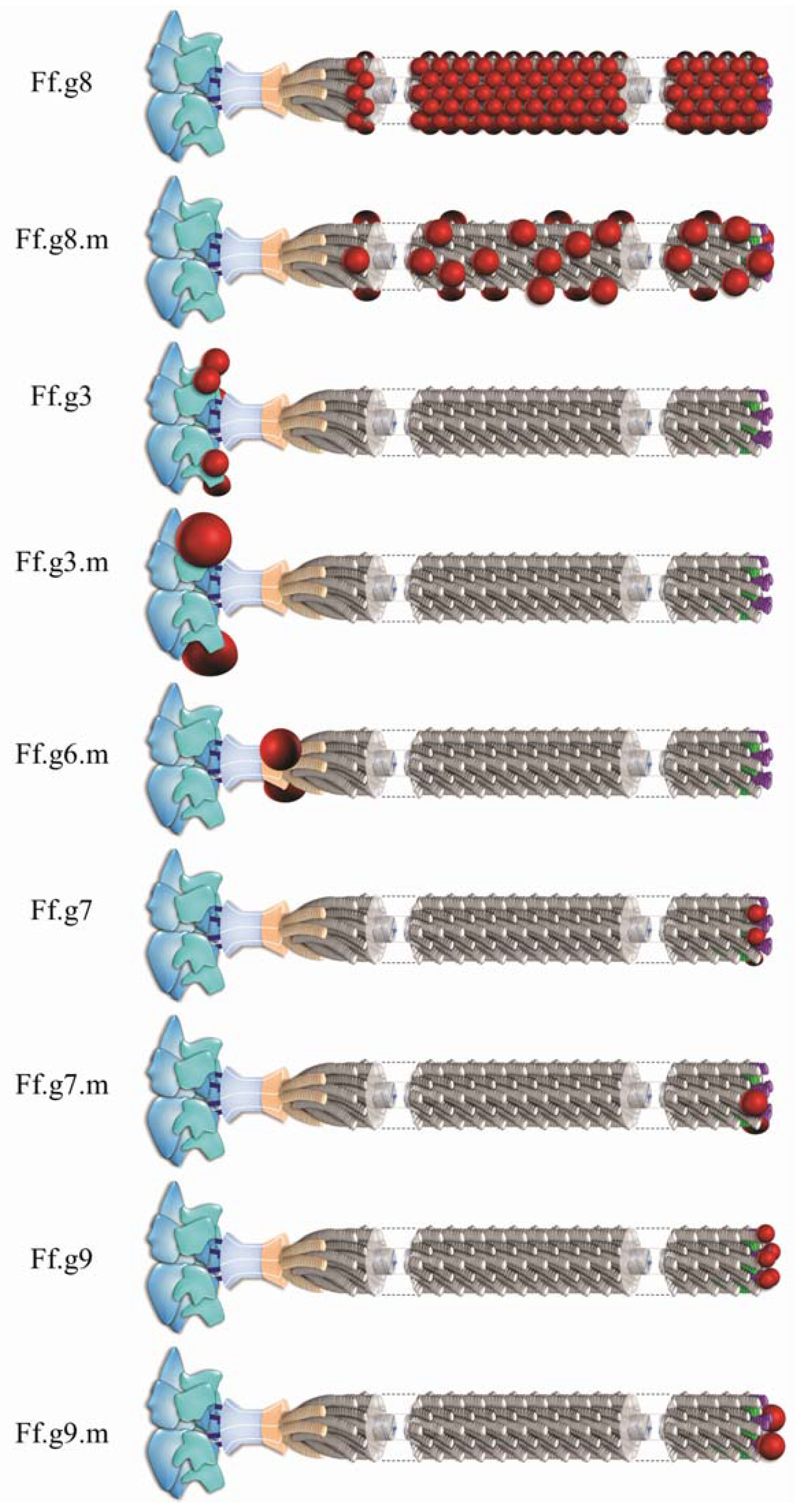
2. Inovirus Associated Vectors



{kind=link}
{kind=link}
{kind=link}
| Pathogens a | Inoviral Vector b | Antigen c | Study Animal Model | Type of Immune Response d | Protection of Vaccinated Animals against Challenge with the Pathogen e | References | |
|---|---|---|---|---|---|---|---|
| Viral | |||||||
| HBV | M13.g8.m | Hepatitis B Surface Antigen | Mice | Specific humoral | ND | Folgori et al., 1994 | [48] |
| HBV | M13.g8.m | Hepatitis B Surface Antigen | Mice | Specific cellular (CD8) | ND | Wan et al., 2001 | [49] |
| HIV-1 | fd.g3 | gp120 (V3 loop) | Rabbits | Specific humoral | ND | Keller et al., 1993 | [50] |
| HIV-1 | M13.g8.m | Unknown | Mice | Specific humoral | ND | Scala et al., 1999 | [51] |
| HIV-1 | fd.g8.m | gp120 (CD4 binding site) | Mice, Rabbits | Non-specific humoral | ND | Zwick et al., 2001 | [52] |
| HIV-1 | M13.g8.m | Unknown | Rhesus macaques | Specific humoral | Partial | Chen et al., 2001 | [53] |
| HIV-1 | fd.g3 | gp120 (CD4 binding site) | Mice | Specific humoral | ND | Dorgham et al., 2005 | [54] |
| HIV-1 | M13.g3 | gp120 (CD4 binding site) | Rabbits | Specific humoral | ND | Wilkinson et al., 2007 | [55] |
| HIV-1 | M13.g3 | Unknown | Mice | Specific humoral | ND | Rodriguez et al., 2007 | [56] |
| HIV-1 | M13.g3 | Unknown | Mice | Specific humoral | ND | Humbert et al., 2007 | [57] |
| HIV-1 | M13.g3 | Unknown | Mice | Specific humoral | ND | Humbert et al., 2008 | [58] |
| HIV-1 | fd.g8.m | Carbohydrates | Rabbits | Non-specific humoral | ND | Menendez et al., 2008 | [59] |
| HIV-1 | M13.g8.m | gp120 (V3 loop) | Mice | Specific cellular (CD8) | ND | Pedrosa-Roldan et al., 2009 | [60] |
| HIV-1 | M13.g8.m | gp120 (V3 loop) | Mice | Specific humoral | ND | Charles-Nino et al., 2011 | [61] |
| HIV-1 | M13.g3 | gp41 (MPER) | Mice, Rabbits | Specific humoral | ND | Rodriguez et al., 2011 | [62] |
| HIV-1 | M13.g3 | gp120 (V3 loop) | Rabbits | Specific humoral | ND | Gazarian et al., 2013 | [63] |
| HPV | fd.g8.m | E7 | Mice | Specific humoral | ND | Lidqvist et al., 2008 | [64] |
| HSV-2 | fd.g8.m | Glycoprotein G2 | Mice | Specific humoral | Significant | Grabowska et al., 2000 | [65] |
| Influenza A | M13.g3 | Hemagglutinin Antigen | Mice | Specific humoral | Significant | Zhong et al., 2011 | [66] |
| Neurotropic Murine Coronavirus | fd.g8.m | Surface Glycoprotein | Mice | Specific humoral | Partial | Yu et al., 2000 | [67] |
| Rabies virus | M13.g3 | Rabies Viral Glycoprotein | Mice | Specific humoral | ND | Houimel et al., 2009 | [68] |
| RSV | fd.g3 | Glycoprotein G | Mice | Specific humoral | Complete | Bastien et al., 1997 | [69] |
| Protozoan | |||||||
| Plasmodium falciparum | fd.g3 | CSP | Mice, Rabbits | Specific humoral | ND | Stoute et al., 1995 | [70] |
| Plasmodium falciparum | F1.g3 | CSP | Mice, Rabbits | Specific humoral | ND | de la Cruz et al., 1988 | [71] |
| Plasmodium falciparum | fd.g8.m | CSP | Rabbits | Specific humoral | ND | Greenwood et al., 1991 | [72] |
| Fungal | |||||||
| Candida albicans | fd.g8.m | Heat Shock Protein 90 | Mice | Specific humoral and cellular (CD4) | Partial | Yang et al., 2005 | [73] |
| Candida albicans | fd.g8.m | Heat Shock Protein 90 | Mice | Specific humoral and cellular (CD4) | Partial | Wang et al., 2006 | [74] |
| Worm | |||||||
| Fasciola hepatica | M13.g3 | Cathepsin L | Sheep | Specific humoral | Partial | Villa-Mancera et al., 2008 | [75] |
| Schistosoma japonicum | M13.g3 | Ferritin of Sj | Mice | Specific humoral | Partial | Tang et al., 2004 | [76] |
| Schistosoma japonicum | M13.g3 | Epitope of mAb SSJ14 | Mice | Specific humoral and cellular (CD4) | Significant | Wang et al., 2006 | [77] |
| Schistosoma japonicum | M13.g3 | Sj338 | Mice | Specific humoral | Partial | Wu et al., 2006 | [78] |
| Taenia solium | M13.g8.m, M13.g3 | GK1, KETc1, KETc7, KETc12 | Guinea Pigs | Non-specific humoral and specific cellular (CD4) | Significant | Manoutcharian et al., 2004 | [79] |
| Trichinella spiralis | M13.g3 | Ts87 | Mice | Specific humoral | Partial | Gu et al., 2008 | [80] |
| Trichirella spiralis | M13.g3 | Ts Paramyosin | Mice | Specific humoral | Partial | Wei et al., 2011 | [81] |
| Diseases | Inoviral Vector a | Antigen b | Study Animal Model | Type of Immune Response c | Protection of Vaccinated Animals against disease progression d | References | |
|---|---|---|---|---|---|---|---|
| Cancer | |||||||
| Colorectal cancer | M13.g3 | EGFR | Mice | Specific humoral | ND | Riemer et al., 2005 A | [82] |
| Melanoma | fd.g8.m | HMW-MAA | Mice | Specific humoral | ND | Riemer et al., 2005 B | [83] |
| Melanoma | fd.g3 | HMW-MAA | Mice | Specific humoral | ND | Luo et al., 2005 | [84] |
| Melanoma | fd.g8.m | MAGE A1 | Mice | Specific cellular (CD8 and CD4) | Significant | Fang et al., 2005 | [85] |
| Melanoma | Ff.g8 or Ff.g8.m | HMW-MAA | Rabbits | Specific humoral | ND | Wagner et al., 2005 | [86] |
| Melanoma | fd.g3 | HMW-MAA | Rabbits | Specific humoral | ND | Luo et al., 2006 | [87] |
| Melanoma | F1.g8.m, M13.g3 | HMW-MAA | Mice, Rabbits | Specific humoral only in mice | ND | Latzka et al., 2011 | [88] |
| Murine mastocytoma P815 | M13.g8.m | P1A | Mice | Specific cellular (CD8 and CD4) | Significant | Wu et al., 2002 | [89] |
| Various types of cancer | M13.g3 | Epitope of mAb BAT | Mice | Specific humoral and cellular (CD8) | Significant | Hardy et al., 2005 | [90] |
| Various types of cancer | M13.g3 | EGFR | Rabbits | Specific humoral | ND | Hartman et al., 2010 | [91] |
| Various types of cancer | M13.g3 | VEGF | Mice | Specific humoral | ND | Li et al., 2013 | [92] |
| Alzheimer’s | |||||||
| fd.g8.m, fd.g3 | EFRH | Guinea pigs | Specific humoral | ND | Frenkel et al., 2000 | [93] | |
| fd.g8.m | EFRH | Mice | Specific humoral | Significant | Frenkel et al., 2003 | [94] | |
| fd.g8.m, fd.g3 | EFRH | Mice | Specific humoral | Partial | Lavie et al., 2004 | [95] | |
| fd.g8.m, fd.g3 | EFRH | Mice | Specific humoral | Partial | Solomon et al., 2007 | [96] |
3. Inovirus Display Technology in Vaccine Design against Non-HIV-1 Diseases
4. HIV-1 Inovirus-Based Vaccines
5. IAVs Induce Strong Cellular Immune Responses
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Day, L.A. Inoviruses. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., Regenmortel, M.H.V.V., Eds.; Academic Press: Oxford, UK, 2008; pp. 117–124. [Google Scholar]
- Marvin, D.A.; Symmons, M.F.; Straus, S.K. Structure and assembly of filamentous bacteriophages. Prog. Biophys. Mol. Biol. 2014, 114, 80–122. [Google Scholar]
- Day, L.A.; Marzec, C.J.; Reisberg, S.A.; Casadevall, A. DNA packing in filamentous bacteriophages. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 509–539. [Google Scholar]
- Rakonjac, J.; Bennett, N.J.; Spagnuolo, J.; Gagic, D.; Russel, M. Filamentous bacteriophage: Biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 2011, 13, 51–76. [Google Scholar]
- Brown, L.R.; Dowell, C.E. Replication of coliphage m-13. I. Effects on host cells after synchronized infection. J. Virol. 1968, 2, 1290–1295. [Google Scholar]
- Marvin, D.A.; Hohn, B. Filamentous bacterial viruses. Bacteriol. Rev. 1969, 33, 172–209. [Google Scholar]
- Beck, E.; Sommer, R.; Auerswald, E.A.; Kurz, C.; Zink, B.; Osterburg, G.; Schaller, H.; Sugimoto, K.; Sugisaki, H.; Okamoto, T.; et al. Nucleotide sequence of bacteriophage fd DNA. Nucleic Acids Res. 1978, 5, 4495–4503. [Google Scholar]
- Hill, D.F.; Petersen, G.B. Nucleotide sequence of bacteriophage f1 DNA. J. Virol. 1982, 44, 32–46. [Google Scholar]
- Van Wezenbeek, P.M.; Hulsebos, T.J.; Schoenmakers, J.G. Nucleotide sequence of the filamentous bacteriophage m13 DNA genome: Comparison with phage fd. Gene 1980, 11, 129–148. [Google Scholar]
- Russel, M.; Model, P. Genetic analysis of the filamentous bacteriophage packaging signal and of the proteins that interact with it. J. Virol. 1989, 63, 3284–3295. [Google Scholar]
- Dotto, G.P.; Enea, V.; Zinder, N.D. Functional analysis of bacteriophage f1 intergenic region. Virology 1981, 114, 463–473. [Google Scholar]
- Bauer, M.; Smith, G.P. Filamentous phage morphogenetic signal sequence and orientation of DNA in the virion and gene-v protein complex. Virology 1988, 167, 166–175. [Google Scholar]
- Shen, C.K.; Ikoku, A.; Hearst, J.E. A specific DNA orientation in the filamentous bacteriophage fd as probed by psoralen crosslinking and electron microscopy. J. Mol. Biol. 1979, 127, 163–175. [Google Scholar]
- Lopez, J.; Webster, R.E. Morphogenesis of filamentous bacteriophage f1: Orientation of extrusion and production of polyphage. Virology 1983, 127, 177–193. [Google Scholar]
- Webster, R.E.; Grant, R.A.; Hamilton, L.A. Orientation of the DNA in the filamentous bacteriophage f1. J. Mol. Biol. 1981, 152, 357–374. [Google Scholar]
- Armstrong, J.; Hewitt, J.A.; Perham, R.N. Chemical modification of the coat protein in bacteriophage fd and orientation of the virion during assembly and disassembly. EMBO J. 1983, 2, 1641–1646. [Google Scholar]
- Smilowitz, H. Bacteriophage f1 infection: Fate of the parental major coat protein. J. Virol. 1974, 13, 94–99. [Google Scholar]
- Trenkner, E.; Bonhoeffer, F.; Gierer, A. The fate of the protein component of bacteriophage fd during infection. Biochem. Biophys. Res. Commun. 1967, 28, 932–939. [Google Scholar]
- Chang, C.N.; Blobel, G.; Model, P. Detection of prokaryotic signal peptidase in an escherichia coli membrane fraction: Endoproteolytic cleavage of nascent f1 pre-coat protein. Proc. Natl. Acad. Sci. USA 1978, 75, 361–365. [Google Scholar]
- Kuhn, A.; Wickner, W. Isolation of mutants in m13 coat protein that affect its synthesis, processing, and assembly into phage. J. Biol. Chem. 1985, 260, 15907–15913. [Google Scholar]
- Kuhn, A.; Wickner, W. Conserved residues of the leader peptide are essential for cleavage by leader peptidase. J. Biol. Chem. 1985, 260, 15914–15918. [Google Scholar]
- Grant, R.A.; Lin, T.C.; Konigsberg, W.; Webster, R.E. Structure of the filamentous bacteriophage-fl—Location of the minor coat protein-a, protein-c and protein-d. J. Biol. Chem. 1981, 256, 539–546. [Google Scholar]
- Makowski, L. Terminating a macromolecular helix. Structural model for the minor proteins of bacteriophage m13. J. Mol. Biol. 1992, 228, 885–892. [Google Scholar]
- Makowski, L. Structural constraints on the display of foreign peptides on filamentous bacteriophages. Gene 1993, 128, 5–11. [Google Scholar]
- Makowski, L. Phage display—Structure, assembly and engineering of filamentous bacteriophage-m13. Curr. Opin. Struct. Biol. 1994, 4, 225–230. [Google Scholar]
- Simons, G.F.M.; Konings, R.N.H.; Schoemakers, J.G.G. Genes-vi, genes-vii, and genes-ix of phage-m13 code for minor capsid proteins of the virion. Proc. Natl. Acad. Sci. USA 1981, 78, 4194–4198. [Google Scholar]
- Rakonjac, J.; Feng, J.; Model, P. Filamentous phage are released from the bacterial membrane by a two-step mechanism involving a short c-terminal fragment of piii. J. Mol. Biol. 1999, 289, 1253–1265. [Google Scholar]
- Caspar, D.L.; Makowski, L. The symmetries of filamentous phage particles. J. Mol. Biol. 1981, 145, 611–617. [Google Scholar]
- Stubbs, G.; Kendall, A. Helical viruses. Adv. Exp. Med. Biol. 2012, 726, 631–658. [Google Scholar]
- Glucksman, M.J.; Bhattacharjee, S.; Makowski, L. Three-dimensional structure of a cloning vector. X-ray diffraction studies of filamentous bacteriophage m13 at 7 a resolution. J. Mol. Biol. 1992, 226, 455–470. [Google Scholar]
- Marzec, C.J.; Day, L.A. A theory of the symmetries of filamentous bacteriophages. Biophys. J. 1988, 53, 425–440. [Google Scholar]
- Kostrikis, L.G.; Reisberg, S.A.; Simon, M.N.; Wall, J.S.; Day, L.A. Export of infectious particles by escherichia coli transfected with the rf DNA of pf1, a virus of pseudomonas aeruginosa strain k. Mol. Microbiol. 1991, 5, 2641–2647. [Google Scholar]
- Wall, J.S.; Hainfeld, J.F.; Simon, M.N. Scanning transmission electron microscopy of nuclear structures. Methods Cell Biol. 1998, 53, 139–164. [Google Scholar]
- Wall, J.S.; Simon, M.N. Scanning transmission electron microscopy of DNA-protein complexes. Methods Mol. Biol. 2001, 148, 589–601. [Google Scholar]
- Marvin, D.A.; Welsh, L.C.; Symmons, M.F.; Scott, W.R.; Straus, S.K. Molecular structure of fd (f1, m13) filamentous bacteriophage refined with respect to x-ray fibre diffraction and solid-state nmr data supports specific models of phage assembly at the bacterial membrane. J. Mol. Biol. 2006, 355, 294–309. [Google Scholar]
- Lubkowski, J.; Hennecke, F.; Pluckthun, A.; Wlodawer, A. The structural basis of phage display elucidated by the crystal structure of the n-terminal domains of g3p. Nat. Struct. Biol. 1998, 5, 140–147. [Google Scholar]
- Smith, G.P.; Scott, J.K. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993, 217, 228–257. [Google Scholar]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar]
- Chasteen, L.; Ayriss, J.; Pavlik, P.; Bradbury, A.R. Eliminating helper phage from phage display. Nucleic Acids Res. 2006, 34, e145. [Google Scholar]
- Kwasnikowski, P.; Kristensen, P.; Markiewicz, W.T. Multivalent display system on filamentous bacteriophage pvii minor coat protein. J. Immunol. Methods 2005, 307, 135–143. [Google Scholar]
- Ploss, M.; Kuhn, A. Membrane insertion and assembly of epitope-tagged gp9 at the tip of the m13 phage. BMC Microbiol. 2011, 11, e211. [Google Scholar]
- Loset, G.A.; Sandlie, I. Next generation phage display by use of pvii and pix as display scaffolds. Methods 2012, 58, 40–46. [Google Scholar]
- Fransen, M.; van Veldhoven, P.P.; Subramani, S. Identification of peroxisomal proteins by using m13 phage protein vi phage display: Molecular evidence that mammalian peroxisomes contain a 2,4-dienoyl-coa reductase. Biochem. J. 1999, 340, 561–568. [Google Scholar]
- Hufton, S.E.; Moerkerk, P.T.; Meulemans, E.V.; de Bruine, A.; Arends, J.W.; Hoogenboom, H.R. Phage display of cdna repertoires: The pvi display system and its applications for the selection of immunogenic ligands. J. Immunol. Methods 1999, 231, 39–51. [Google Scholar]
- Jespers, L.S.; Messens, J.H.; de Keyser, A.; Eeckhout, D.; van den Brande, I.; Gansemans, Y.G.; Lauwereys, M.J.; Vlasuk, G.P.; Stanssens, P.E. Surface expression and ligand-based selection of cdnas fused to filamentous phage gene vi. Biotechnology (NY) 1995, 13, 378–382. [Google Scholar]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar]
- Tikunova, N.V.; Morozova, V.V. Phage display on the base of filamentous bacteriophages: Application for recombinant antibodies selection. Acta Naturae 2009, 1, 20–28. [Google Scholar]
- Folgori, A.; Tafi, R.; Meola, A.; Felici, F.; Galfre, G.; Cortese, R.; Monaci, P.; Nicosia, A. A general strategy to identify mimotopes of pathological antigens using only random peptide libraries and human sera. EMBO J. 1994, 13, 2236–2243. [Google Scholar]
- Wan, Y.; Wu, Y.; Bian, J.; Wang, X.Z.; Zhou, W.; Jia, Z.C.; Tan, Y.; Zhou, L. Induction of hepatitis b virus-specific cytotoxic t lymphocytes response in vivo by filamentous phage display vaccine. Vaccine 2001, 19, 2918–2923. [Google Scholar]
- Keller, P.M.; Arnold, B.A.; Shaw, A.R.; Tolman, R.L.; van Middlesworth, F.; Bondy, S.; Rusiecki, V.K.; Koenig, S.; Zolla-Pazner, S.; Conard, P.; et al. Identification of HIV vaccine candidate peptides by screening random phage epitope libraries. Virology 1993, 193, 709–716. [Google Scholar]
- Scala, G.; Chen, X.; Liu, W.; Telles, J.N.; Cohen, O.J.; Vaccarezza, M.; Igarashi, T.; Fauci, A.S. Selection of hiv-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. J. Immunol. 1999, 162, 6155–6161. [Google Scholar]
- Zwick, M.B.; Bonnycastle, L.L.; Menendez, A.; Irving, M.B.; Barbas, C.F., 3rd; Parren, P.W.; Burton, D.R.; Scott, J.K. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J. Virol. 2001, 75, 6692–6699. [Google Scholar]
- Chen, X.; Scala, G.; Quinto, I.; Liu, W.; Chun, T.W.; Justement, J.S.; Cohen, O.J.; vanCott, T.C.; Iwanicki, M.; Lewis, M.G.; et al. Protection of rhesus macaques against disease progression from pathogenic shiv-89.6pd by vaccination with phage-displayed hiv-1 epitopes. Nat. Med. 2001, 7, 1225–1231. [Google Scholar]
- Dorgham, K.; Dogan, I.; Bitton, N.; Parizot, C.; Cardona, V.; Debre, P.; Hartley, O.; Gorochov, G. Immunogenicity of HIV type 1 gp120 cd4 binding site phage mimotopes. AIDS Res. Hum. Retrovir. 2005, 21, 82–92. [Google Scholar]
- Wilkinson, R.A.; Evans, J.R.; Jacobs, J.M.; Slunaker, D.; Pincus, S.H.; Pinter, A.; Parkos, C.A.; Burritt, J.B.; Teintze, M. Peptides selected from a phage display library with an hiv-neutralizing antibody elicit antibodies to hiv gp120 in rabbits, but not to the same epitope. AIDS Res. Hum. Retrovir. 2007, 23, 1416–1427. [Google Scholar]
- Palacios-Rodriguez, Y.; Gazarian, T.; Rowley, M.; Majluf-Cruz, A.; Gazarian, K. Collection of phage-peptide probes for hiv-1 immunodominant loop-epitope. J. Microbiol. Methods 2007, 68, 225–235. [Google Scholar]
- Humbert, M.; Antoni, S.; Brill, B.; Landersz, M.; Rodes, B.; Soriano, V.; Wintergerst, U.; Knechten, H.; Staszewski, S.; von Laer, D.; et al. Mimotopes selected with antibodies from HIV-1-neutralizing long-term non-progressor plasma. Eur. J. Immunol. 2007, 37, 501–515. [Google Scholar]
- Humbert, M.; Rasmussen, R.A.; Ong, H.; Kaiser, F.M.; Hu, S.L.; Ruprecht, R.M. Inducing cross-clade neutralizing antibodies against hiv-1 by immunofocusing. PLoS One 2008, 3, e3937. [Google Scholar]
- Menendez, A.; Calarese, D.A.; Stanfield, R.L.; Chow, K.C.; Scanlan, C.N.; Kunert, R.; Katinger, H.; Burton, D.R.; Wilson, I.A.; Scott, J.K. A peptide inhibitor of hiv-1 neutralizing antibody 2g12 is not a structural mimic of the natural carbohydrate epitope on gp120. FASEB J. 2008, 22, 1380–1392. [Google Scholar]
- Pedroza-Roldan, C.; Charles-Nino, C.; Saavedra, R.; Govezensky, T.; Vaca, L.; Avaniss-Aghajani, E.; Gevorkian, G.; Manoutcharian, K. Variable epitope library-based vaccines: Shooting moving targets. Mol. Immunol. 2009, 47, 270–282. [Google Scholar]
- Charles-Nino, C.; Pedroza-Roldan, C.; Viveros, M.; Gevorkian, G.; Manoutcharian, K. Variable epitope libraries: New vaccine immunogens capable of inducing broad human immunodeficiency virus type 1-neutralizing antibody response. Vaccine 2011, 29, 5313–5321. [Google Scholar]
- Palacios-Rodriguez, Y.; Gazarian, T.; Huerta, L.; Gazarian, K. Constrained peptide models from phage display libraries highlighting the cognate epitope-specific potential of the anti-HIV-1 mab 2f5. Immunol. Lett. 2011, 136, 80–89. [Google Scholar]
- Gazarian, K.G.; Palacios-Rodriguez, Y.; Gazarian, T.G.; Huerta, L. Hiv-1 v3 loop crown epitope-focused mimotope selection by patient serum from random phage display libraries: Implications for the epitope structural features. Mol. Immunol. 2013, 54, 148–156. [Google Scholar]
- Lidqvist, M.; Nilsson, O.; Holmgren, J.; Hall, C.; Fermer, C. Phage display for site-specific immunization and characterization of high-risk human papillomavirus specific e7 monoclonal antibodies. J. Immunol. Methods 2008, 337, 88–96. [Google Scholar]
- Grabowska, A.M.; Jennings, R.; Laing, P.; Darsley, M.; Jameson, C.L.; Swift, L.; Irving, W.L. Immunisation with phage displaying peptides representing single epitopes of the glycoprotein g can give rise to partial protective immunity to HSV-2. Virology 2000, 269, 47–53. [Google Scholar]
- Zhong, Y.; Cai, J.; Zhang, C.; Xing, X.; Qin, E.; He, J.; Mao, P.; Cheng, J.; Liu, K.; Xu, D.; et al. Mimotopes selected with neutralizing antibodies against multiple subtypes of influenza A. Virol. J. 2011, 8, e542. [Google Scholar]
- Yu, M.W.; Scott, J.K.; Fournier, A.; Talbot, P.J. Characterization of murine coronavirus neutralization epitopes with phage-displayed peptides. Virology 2000, 271, 182–196. [Google Scholar]
- Houimel, M.; Dellagi, K. Peptide mimotopes of rabies virus glycoprotein with immunogenic activity. Vaccine 2009, 27, 4648–4655. [Google Scholar]
- Bastien, N.; Trudel, M.; Simard, C. Protective immune responses induced by the immunization of mice with a recombinant bacteriophage displaying an epitope of the human respiratory syncytial virus. Virology 1997, 234, 118–122. [Google Scholar]
- Stoute, J.A.; Ballou, W.R.; Kolodny, N.; Deal, C.D.; Wirtz, R.A.; Lindler, L.E. Induction of humoral immune response against plasmodium falciparum sporozoites by immunization with a synthetic peptide mimotope whose sequence was derived from screening a filamentous phage epitope library. Infect. Immun. 1995, 63, 934–939. [Google Scholar]
- De la Cruz, V.F.; Lal, A.A.; McCutchan, T.F. Immunogenicity and epitope mapping of foreign sequences via genetically engineered filamentous phage. J. Biol. Chem. 1988, 263, 4318–4322. [Google Scholar]
- Greenwood, J.; Willis, A.E.; Perham, R.N. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from plasmodium falciparum circumsporozoite protein as antigens. J. Mol. Biol. 1991, 220, 821–827. [Google Scholar]
- Yang, Q.; Wang, L.; Lu, D.N.; Gao, R.J.; Song, J.N.; Hua, P.Y.; Yuan, D.W. Prophylactic vaccination with phage-displayed epitope of c. Albicans elicits protective immune responses against systemic candidiasis in c57bl/6 mice. Vaccine 2005, 23, 4088–4096. [Google Scholar]
- Wang, G.; Sun, M.; Fang, J.; Yang, Q.; Tong, H.; Wang, L. Protective immune responses against systemic candidiasis mediated by phage-displayed specific epitope of candida albicans heat shock protein 90 in c57bl/6j mice. Vaccine 2006, 24, 6065–6073. [Google Scholar]
- Villa-Mancera, A.; Quiroz-Romero, H.; Correa, D.; Ibarra, F.; Reyes-Perez, M.; Reyes-Vivas, H.; Lopez-Velazquez, G.; Gazarian, K.; Gazarian, T.; Alonso, R.A. Induction of immunity in sheep to fasciola hepatica with mimotopes of cathepsin l selected from a phage display library. Parasitology 2008, 135, 1437–1445. [Google Scholar]
- Tang, L.F.; Yi, X.Y.; Zeng, X.F.; Wang, L.Q.; Zhang, S.K. Schistosoma japonicum: Isolation and identification of peptides mimicking ferritin epitopes from phage display library. Acta Biochim. Biophys. Sin. (Shanghai) 2004, 36, 206–210. [Google Scholar]
- Wang, X.Z.; Fu, Z.Q.; Huang, S.P.; Zhu, G.Q.; Cai, Y.M.; Lin, J.J. Studies on phage displayed mimotopes of a protective monoclonal antibody (ssj14) against Schistosoma japonicum. Sheng Wu Gong Cheng Xue Bao 2006, 22, 119–124. [Google Scholar]
- Wu, H.W.; Hu, X.M.; Wang, Y.; Kurtis, J.D.; Zeng, F.J.; McGarvey, S.T.; Wu, G.L.; Zhang, Z.S.; Hua, Z.C. Protective immunity induced by phage displayed mitochondrial related peptides of schistosoma japonicum. Acta Trop. 2006, 99, 200–207. [Google Scholar]
- Manoutcharian, K.; Diaz-Orea, A.; Gevorkian, G.; Fragoso, G.; Acero, G.; Gonzalez, E.; de Aluja, A.; Villalobos, N.; Gomez-Conde, E.; Sciutto, E. Recombinant bacteriophage-based multiepitope vaccine against taenia solium pig cysticercosis. Vet. Immunol. Immunopathol. 2004, 99, 11–24. [Google Scholar]
- Gu, Y.; Li, J.; Zhu, X.; Yang, J.; Li, Q.; Liu, Z.; Yu, S.; Li, Y. Trichinella spiralis: Characterization of phage-displayed specific epitopes and their protective immunity in balb/c mice. Exp. Parasitol. 2008, 118, 66–74. [Google Scholar]
- Wei, J.; Gu, Y.; Yang, J.; Yang, Y.; Wang, S.; Cui, S.; Zhu, X. Identification and characterization of protective epitope of trichinella spiralis paramyosin. Vaccine 2011, 29, 3162–3168. [Google Scholar]
- Riemer, A.B.; Kurz, H.; Klinger, M.; Scheiner, O.; Zielinski, C.C.; Jensen-Jarolim, E. Vaccination with cetuximab mimotopes and biological properties of induced anti-epidermal growth factor receptor antibodies. J. Natl. Cancer Inst. 2005, 97, 1663–1670. [Google Scholar]
- Riemer, A.B.; Hantusch, B.; Sponer, B.; Kraml, G.; Hafner, C.; Zielinski, C.C.; Scheiner, O.; Pehamberger, H.; Jensen-Jarolim, E. High-molecular-weight melanoma-associated antigen mimotope immunizations induce antibodies recognizing melanoma cells. Cancer Immunol. Immunother. 2005, 54, 677–684. [Google Scholar]
- Luo, W.; Hsu, J.C.; Tsao, C.Y.; Ko, E.; Wang, X.; Ferrone, S. Differential immunogenicity of two peptides isolated by high molecular weight-melanoma-associated antigen-specific monoclonal antibodies with different affinities. J. Immunol. 2005, 174, 7104–7110. [Google Scholar]
- Fang, J.; Wang, G.; Yang, Q.; Song, J.; Wang, Y.; Wang, L. The potential of phage display virions expressing malignant tumor specific antigen mage-a1 epitope in murine model. Vaccine 2005, 23, 4860–4866. [Google Scholar]
- Wagner, S.; Hafner, C.; Allwardt, D.; Jasinska, J.; Ferrone, S.; Zielinski, C.C.; Scheiner, O.; Wiedermann, U.; Pehamberger, H.; Breiteneder, H. Vaccination with a human high molecular weight melanoma-associated antigen mimotope induces a humoral response inhibiting melanoma cell growth in vitro. J. Immunol. 2005, 174, 976–982. [Google Scholar]
- Luo, W.; Ko, E.; Hsu, J.C.; Wang, X.; Ferrone, S. Targeting melanoma cells with human high molecular weight-melanoma associated antigen-specific antibodies elicited by a peptide mimotope: Functional effects. J. Immunol. 2006, 176, 6046–6054. [Google Scholar]
- Latzka, J.; Gaier, S.; Hofstetter, G.; Balazs, N.; Smole, U.; Ferrone, S.; Scheiner, O.; Breiteneder, H.; Pehamberger, H.; Wagner, S. Specificity of mimotope-induced anti-high molecular weight-melanoma associated antigen (hmw-maa) antibodies does not ensure biological activity. PLoS One 2011, 6, e19383. [Google Scholar]
- Wu, Y.; Wan, Y.; Bian, J.; Zhao, J.; Jia, Z.; Zhou, L.; Zhou, W.; Tan, Y. Phage display particles expressing tumor-specific antigens induce preventive and therapeutic anti-tumor immunity in murine p815 model. Int. J. Cancer 2002, 98, 748–753. [Google Scholar]
- Hardy, B.; Raiter, A. A mimotope peptide-based anti-cancer vaccine selected by bat monoclonal antibody. Vaccine 2005, 23, 4283–4291. [Google Scholar]
- Hartmann, C.; Muller, N.; Blaukat, A.; Koch, J.; Benhar, I.; Wels, W.S. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-egfr immune response. Oncogene 2010, 29, 4517–4527. [Google Scholar]
- Li, W.; Ran, Y.; Li, M.; Zhang, K.; Qin, X.; Xue, X.; Zhang, C.; Hao, Q.; Zhang, W.; Zhang, Y. Mimotope vaccination for epitope-specific induction of anti-vegf antibodies. BMC Biotechnol. 2013, 13, e77. [Google Scholar]
- Frenkel, D.; Katz, O.; Solomon, B. Immunization against alzheimer’s beta -amyloid plaques via efrh phage administration. Proc. Natl. Acad. Sci. USA 2000, 97, 11455–11459. [Google Scholar]
- Frenkel, D.; Dewachter, I.; van Leuven, F.; Solomon, B. Reduction of beta-amyloid plaques in brain of transgenic mouse model of alzheimer’s disease by efrh-phage immunization. Vaccine 2003, 21, 1060–1065. [Google Scholar]
- Lavie, V.; Becker, M.; Cohen-Kupiec, R.; Yacoby, I.; Koppel, R.; Wedenig, M.; Hutter-Paier, B.; Solomon, B. Efrh-phage immunization of alzheimer’s disease animal model improves behavioral performance in morris water maze trials. J. Mol. Neurosci. 2004, 24, 105–113. [Google Scholar]
- Solomon, B. Active immunization against alzheimer’s beta-amyloid peptide using phage display technology. Vaccine 2007, 25, 3053–3056. [Google Scholar]
- Frenkel, D.; Balass, M.; Solomon, B. N-terminal efrh sequence of alzheimer’s beta-amyloid peptide represents the epitope of its anti-aggregating antibodies. J. Neuroimmunol. 1998, 88, 85–90. [Google Scholar]
- Lo Passo, C.; Romeo, A.; Pernice, I.; Donato, P.; Midiri, A.; Mancuso, G.; Arigo, M.; Biondo, C.; Galbo, R.; Papasergi, S.; et al. Peptide mimics of the group b meningococcal capsule induce bactericidal and protective antibodies after immunization. J. Immunol. 2007, 178, 4417–4423. [Google Scholar]
- Irving, M.B.; Pan, O.; Scott, J.K. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr. Opin. Chem. Biol. 2001, 5, 314–324. [Google Scholar]
- Henry, K.A.; Murira, A.; van Houten, N.E.; Scott, J.K. Developing strategies to enhance and focus humoral immune responses using filamentous phage as a model antigen. Bioeng. Bugs 2011, 2, 275–283. [Google Scholar]
- Morales, J.; Martinez, J.J.; Manoutcharian, K.; Hernandez, M.; Fleury, A.; Gevorkian, G.; Acero, G.; Blancas, A.; Toledo, A.; Cervantes, J.; et al. Inexpensive anti-cysticercosis vaccine: S3pvac expressed in heat inactivated m13 filamentous phage proves effective against naturally acquired taenia solium porcine cysticercosis. Vaccine 2008, 26, 2899–2905. [Google Scholar]
- Muster, T.; Steindl, F.; Purtscher, M.; Trkola, A.; Klima, A.; Himmler, G.; Ruker, F.; Katinger, H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 1993, 67, 6642–6647. [Google Scholar]
- Buchacher, A.; Predl, R.; Strutzenberger, K.; Steinfellner, W.; Trkola, A.; Purtscher, M.; Gruber, G.; Tauer, C.; Steindl, F.; Jungbauer, A.; et al. Generation of human monoclonal antibodies against hiv-1 proteins; electrofusion and epstein-barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res. Hum. Retrovir. 1994, 10, 359–369. [Google Scholar]
- Barbas, C.F., 3rd; Hu, D.; Dunlop, N.; Sawyer, L.; Cababa, D.; Hendry, R.M.; Nara, P.L.; Burton, D.R. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. USA 1994, 91, 3809–3813. [Google Scholar]
- Mascola, J.R.; Haynes, B.F. Hiv-1 neutralizing antibodies: Understanding nature’s pathways. Immunol. Rev. 2013, 254, 225–244. [Google Scholar]
- Klein, F.; Mouquet, H.; Dosenovic, P.; Scheid, J.F.; Scharf, L.; Nussenzweig, M.C. Antibodies in HIV-1 vaccine development and therapy. Science 2013, 341, 1199–1204. [Google Scholar]
- Kwong, P.D.; Mascola, J.R.; Nabel, G.J. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: The end of the beginning. Nat. Rev. Immunol. 2013, 13, 693–701. [Google Scholar]
- Binley, J.M.; Wrin, T.; Korber, B.; Zwick, M.B.; Wang, M.; Chappey, C.; Stiegler, G.; Kunert, R.; Zolla-Pazner, S.; Katinger, H.; et al. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 2004, 78, 13232–13252. [Google Scholar]
- Trkola, A.; Pomales, A.B.; Yuan, H.; Korber, B.; Maddon, P.J.; Allaway, G.P.; Katinger, H.; Barbas, C.F., 3rd; Burton, D.R.; Ho, D.D.; et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric cd4-igg. J. Virol. 1995, 69, 6609–6617. [Google Scholar]
- Ofek, G.; Tang, M.; Sambor, A.; Katinger, H.; Mascola, J.R.; Wyatt, R.; Kwong, P.D. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2f5 in complex with its gp41 epitope. J. Virol. 2004, 78, 10724–10737. [Google Scholar]
- Cardoso, R.M.; Zwick, M.B.; Stanfield, R.L.; Kunert, R.; Binley, J.M.; Katinger, H.; Burton, D.R.; Wilson, I.A. Broadly neutralizing anti-hiv antibody 4e10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 2005, 22, 163–173. [Google Scholar]
- Zwick, M.B.; Labrijn, A.F.; Wang, M.; Spenlehauer, C.; Saphire, E.O.; Binley, J.M.; Moore, J.P.; Stiegler, G.; Katinger, H.; Burton, D.R.; et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 2001, 75, 10892–10905. [Google Scholar]
- Zhu, Z.; Qin, H.R.; Chen, W.; Zhao, Q.; Shen, X.; Schutte, R.; Wang, Y.; Ofek, G.; Streaker, E.; Prabakaran, P.; et al. Cross-reactive hiv-1-neutralizing human monoclonal antibodies identified from a patient with 2f5-like antibodies. J. Virol. 2011, 85, 11401–11408. [Google Scholar]
- Morris, L.; Chen, X.; Alam, M.; Tomaras, G.; Zhang, R.; Marshall, D.J.; Chen, B.; Parks, R.; Foulger, A.; Jaeger, F.; et al. Isolation of a human anti-hiv gp41 membrane proximal region neutralizing antibody by antigen-specific single b cell sorting. PLoS One 2011, 6, e23532. [Google Scholar]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an african donor reveal a new hiv-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar]
- Bonsignori, M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; Morris, L.; Gray, E.; Marshall, D.J.; Crump, J.A.; Kapiga, S.H.; Sam, N.E.; et al. Analysis of a clonal lineage of hiv-1 envelope v2/v3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 2011, 85, 9998–10009. [Google Scholar]
- Bonsignori, M.; Montefiori, D.C.; Wu, X.; Chen, X.; Hwang, K.K.; Tsao, C.Y.; Kozink, D.M.; Parks, R.J.; Tomaras, G.D.; Crump, J.A.; et al. Two distinct broadly neutralizing antibody specificities of different clonal lineages in a single hiv-1-infected donor: Implications for vaccine design. J. Virol. 2012, 86, 4688–4692. [Google Scholar]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.P.; Wang, S.K.; Ramos, A.; Chan-Hui, P.Y.; Moyle, M.; et al. Broad neutralization coverage of hiv by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar]
- Burton, D.R.; Pyati, J.; Koduri, R.; Sharp, S.J.; Thornton, G.B.; Parren, P.W.; Sawyer, L.S.; Hendry, R.M.; Dunlop, N.; Nara, P.L.; et al. Efficient neutralization of primary isolates of hiv-1 by a recombinant human monoclonal antibody. Science 1994, 266, 1024–1027. [Google Scholar]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van Ryk, D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; et al. Structural definition of a conserved neutralization epitope on hiv-1 gp120. Nature 2007, 445, 732–737. [Google Scholar]
- Corti, D.; Langedijk, J.P.; Hinz, A.; Seaman, M.S.; Vanzetta, F.; Fernandez-Rodriguez, B.M.; Silacci, C.; Pinna, D.; Jarrossay, D.; Balla-Jhagjhoorsingh, S.; et al. Analysis of memory b cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One 2010, 5, e8805. [Google Scholar]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing hiv-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar]
- Wu, X.; Yang, Z.Y.; Li, Y.; Hogerkorp, C.M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to hiv-1. Science 2010, 329, 856–861. [Google Scholar]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of hiv-1 by antibody vrc01. Science 2010, 329, 811–817. [Google Scholar]
- Wu, X.; Zhou, T.; Zhu, J.; Zhang, B.; Georgiev, I.; Wang, C.; Chen, X.; Longo, N.S.; Louder, M.; McKee, K.; et al. Focused evolution of hiv-1 neutralizing antibodies revealed by structures and deep sequencing. Science 2011, 333, 1593–1602. [Google Scholar]
- Scheid, J.F.; Mouquet, H.; Ueberheide, B.; Diskin, R.; Klein, F.; Oliveira, T.Y.; Pietzsch, J.; Fenyo, D.; Abadir, A.; Velinzon, K.; et al. Sequence and structural convergence of broad and potent hiv antibodies that mimic cd4 binding. Science 2011, 333, 1633–1637. [Google Scholar]
- Diskin, R.; Scheid, J.F.; Marcovecchio, P.M.; West, A.P., Jr.; Klein, F.; Gao, H.; Gnanapragasam, P.N.; Abadir, A.; Seaman, M.S.; Nussenzweig, M.C.; et al. Increasing the potency and breadth of an hiv antibody by using structure-based rational design. Science 2011, 334, 1289–1293. [Google Scholar]
- Calarese, D.A.; Scanlan, C.N.; Zwick, M.B.; Deechongkit, S.; Mimura, Y.; Kunert, R.; Zhu, P.; Wormald, M.R.; Stanfield, R.L.; Roux, K.H.; et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 2003, 300, 2065–2071. [Google Scholar]
- Hassapis, K.A.; Kostrikis, L.G. Hiv-1 vaccine strategies utilizing viral vectors including antigen- displayed inoviral vectors. Curr. HIV Res. 2013, 11, 610–622. [Google Scholar]
- Cecilia, D.; Kleeberger, C.; Munoz, A.; Giorgi, J.V.; Zolla-Pazner, S. A longitudinal study of neutralizing antibodies and disease progression in hiv-1-infected subjects. J. Infect. Dis. 1999, 179, 1365–1374. [Google Scholar]
- Montefiori, D.C.; Pantaleo, G.; Fink, L.M.; Zhou, J.T.; Zhou, J.Y.; Bilska, M.; Miralles, G.D.; Fauci, A.S. Neutralizing and infection-enhancing antibody responses to human immunodeficiency virus type 1 in long-term nonprogressors. J. Infect. Dis. 1996, 173, 60–67. [Google Scholar]
- Pilgrim, A.K.; Pantaleo, G.; Cohen, O.J.; Fink, L.M.; Zhou, J.Y.; Zhou, J.T.; Bolognesi, D.P.; Fauci, A.S.; Montefiori, D.C. Neutralizing antibody responses to human immunodeficiency virus type 1 in primary infection and long-term-nonprogressive infection. J. Infect. Dis. 1997, 176, 924–932. [Google Scholar]
- Wang, Z.; Li, T.Y.; Li, J.Y.; Chen, L.L.; Liu, Y.J.; Li, H.P.; Bao, Z.Y.; Wang, X.L.; Zhuang, D.M.; Liu, S.Y.; et al. Similar neutralizing activity in the hiv-1 infected long term non-progressors(ltnps) and typical progressors(tps). Virol. Sin. 2012, 27, 165–171. [Google Scholar]
- Haynes, B.F.; Fleming, J.; St Clair, E.W.; Katinger, H.; Stiegler, G.; Kunert, R.; Robinson, J.; Scearce, R.M.; Plonk, K.; Staats, H.F.; et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing hiv-1 antibodies. Science 2005, 308, 1906–1908. [Google Scholar]
- Alam, S.M.; McAdams, M.; Boren, D.; Rak, M.; Scearce, R.M.; Gao, F.; Camacho, Z.T.; Gewirth, D.; Kelsoe, G.; Chen, P.; et al. The role of antibody polyspecificity and lipid reactivity in binding of broadly neutralizing anti-hiv-1 envelope human monoclonal antibodies 2f5 and 4e10 to glycoprotein 41 membrane proximal envelope epitopes. J. Immunol. 2007, 178, 4424–4435. [Google Scholar]
- Sanchez-Martinez, S.; Lorizate, M.; Hermann, K.; Kunert, R.; Basanez, G.; Nieva, J.L. Specific phospholipid recognition by human immunodeficiency virus type-1 neutralizing anti-gp41 2f5 antibody. FEBS Lett. 2006, 580, 2395–2399. [Google Scholar]
- Scanlan, C.N.; Pantophlet, R.; Wormald, M.R.; Ollmann Saphire, E.; Stanfield, R.; Wilson, I.A.; Katinger, H.; Dwek, R.A.; Rudd, P.M.; Burton, D.R. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2g12 recognizes a cluster of alpha1-->2 mannose residues on the outer face of gp120. J. Virol. 2002, 76, 7306–7321. [Google Scholar]
- Haynes, B.F.; Moody, M.A.; Verkoczy, L.; Kelsoe, G.; Alam, S.M. Antibody polyspecificity and neutralization of hiv-1: A hypothesis. Hum. Antibodies 2005, 14, 59–67. [Google Scholar]
- Verkoczy, L.; Diaz, M.; Holl, T.M.; Ouyang, Y.B.; Bouton-Verville, H.; Alam, S.M.; Liao, H.X.; Kelsoe, G.; Haynes, B.F. Autoreactivity in an hiv-1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc. Natl. Acad. Sci. USA 2010, 107, 181–186. [Google Scholar]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic siv by an effector memory t-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar]
- Hansen, S.G.; Piatak, M., Jr.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic siv infection. Nature 2013, 502, 100–104. [Google Scholar]
- De Berardinis, P.; Sartorius, R.; Fanutti, C.; Perham, R.N.; del Pozzo, G.; Guardiola, J. Phage display of peptide epitopes from hiv-1 elicits strong cytolytic responses. Nat. Biotechnol. 2000, 18, 873–876. [Google Scholar]
- Gaubin, M.; Fanutti, C.; Mishal, Z.; Durrbach, A.; de Berardinis, P.; Sartorius, R.; del Pozzo, G.; Guardiola, J.; Perham, R.N.; Piatier-Tonneau, D. Processing of filamentous bacteriophage virions in antigen-presenting cells targets both hla class i and class ii peptide loading compartments. DNA Cell Biol. 2003, 22, 11–18. [Google Scholar]
- Sartorius, R.; Bettua, C.; D’Apice, L.; Caivano, A.; Trovato, M.; Russo, D.; Zanoni, I.; Granucci, F.; Mascolo, D.; Barba, P.; et al. Vaccination with filamentous bacteriophages targeting dec-205 induces dc maturation and potent anti-tumor t-cell responses in the absence of adjuvants. Eur. J. Immunol. 2011, 41, 2573–2584. [Google Scholar]
- McMichael, A.J.; Koff, W.C. Vaccines that stimulate t cell immunity to hiv-1: The next step. Nat. Immunol. 2014, 15, 319–322. [Google Scholar]
- Koup, R.A.; Douek, D.C. Vaccine design for cd8 t lymphocyte responses. Cold Spring Harb. Perspect. Med. 2011, 1, a007252. [Google Scholar]
- Barouch, D.H. Novel adenovirus vector-based vaccines for hiv-1. Curr. Opin. HIV AIDS 2010, 5, 386–390. [Google Scholar]
- Ross, A.L.; Brave, A.; Scarlatti, G.; Manrique, A.; Buonaguro, L. Progress towards development of an hiv vaccine: Report of the aids vaccine 2009 conference. Lancet Infect. Dis. 2010, 10, 305–316. [Google Scholar]
- Cohen, Y.Z.; Dolin, R. Novel hiv vaccine strategies: Overview and perspective. Ther. Adv. Vaccines 2013, 1, 99–112. [Google Scholar]
- O’Connell, R.J.; Kim, J.H.; Corey, L.; Michael, N.L. Human immunodeficiency virus vaccine trials. Cold Spring Harb. Perspect. Med. 2012, 2, a007351. [Google Scholar]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity hiv-1 vaccine (the step study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with alvac and aidsvax to prevent hiv-1 infection in thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar]
- Johnson, J.A.; Barouch, D.H.; Baden, L.R. Nonreplicating vectors in hiv vaccines. Curr. Opin. HIV AIDS 2013, 8, 412–420. [Google Scholar]
- Parks, C.L.; Picker, L.J.; King, C.R. Development of replication-competent viral vectors for hiv vaccine delivery. Curr. Opin. HIV AIDS 2013, 8, 402–411. [Google Scholar]
- Russel, M. Filamentous phage assembly. Mol. Microbiol. 1991, 5, 1607–1613. [Google Scholar]
- Date, T.; Wickner, W. Isolation of the escherichia coli leader peptidase gene and effects of leader peptidase overproduction in vivo. Proc. Natl. Acad. Sci. USA 1981, 78, 6106–6110. [Google Scholar]
- Krag, D.N.; Shukla, G.S.; Shen, G.P.; Pero, S.; Ashikaga, T.; Fuller, S.; Weaver, D.L.; Burdette-Radoux, S.; Thomas, C. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006, 66, 7724–7733. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassapis, K.A.; Stylianou, D.C.; Kostrikis, L.G. Architectural Insight into Inovirus-Associated Vectors (IAVs) and Development of IAV-Based Vaccines Inducing Humoral and Cellular Responses: Implications in HIV-1 Vaccines. Viruses 2014, 6, 5047-5076. https://doi.org/10.3390/v6125047
Hassapis KA, Stylianou DC, Kostrikis LG. Architectural Insight into Inovirus-Associated Vectors (IAVs) and Development of IAV-Based Vaccines Inducing Humoral and Cellular Responses: Implications in HIV-1 Vaccines. Viruses. 2014; 6(12):5047-5076. https://doi.org/10.3390/v6125047
Chicago/Turabian StyleHassapis, Kyriakos A., Dora C. Stylianou, and Leondios G. Kostrikis. 2014. "Architectural Insight into Inovirus-Associated Vectors (IAVs) and Development of IAV-Based Vaccines Inducing Humoral and Cellular Responses: Implications in HIV-1 Vaccines" Viruses 6, no. 12: 5047-5076. https://doi.org/10.3390/v6125047
APA StyleHassapis, K. A., Stylianou, D. C., & Kostrikis, L. G. (2014). Architectural Insight into Inovirus-Associated Vectors (IAVs) and Development of IAV-Based Vaccines Inducing Humoral and Cellular Responses: Implications in HIV-1 Vaccines. Viruses, 6(12), 5047-5076. https://doi.org/10.3390/v6125047