CD81 and Hepatitis C Virus (HCV) Infection

Abstract
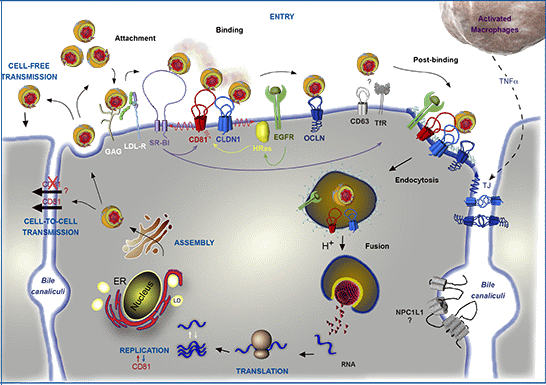
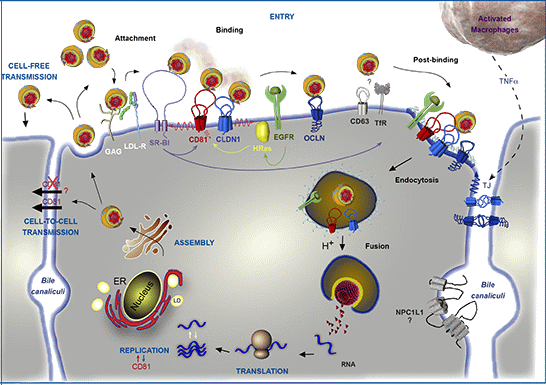
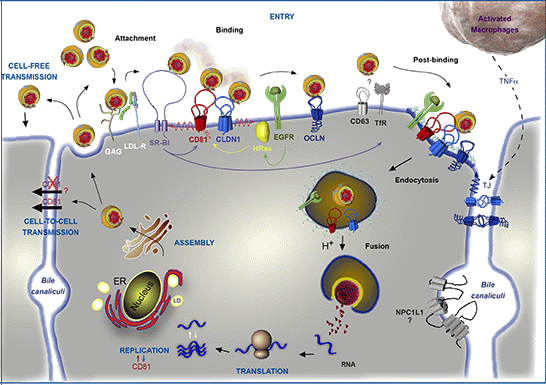
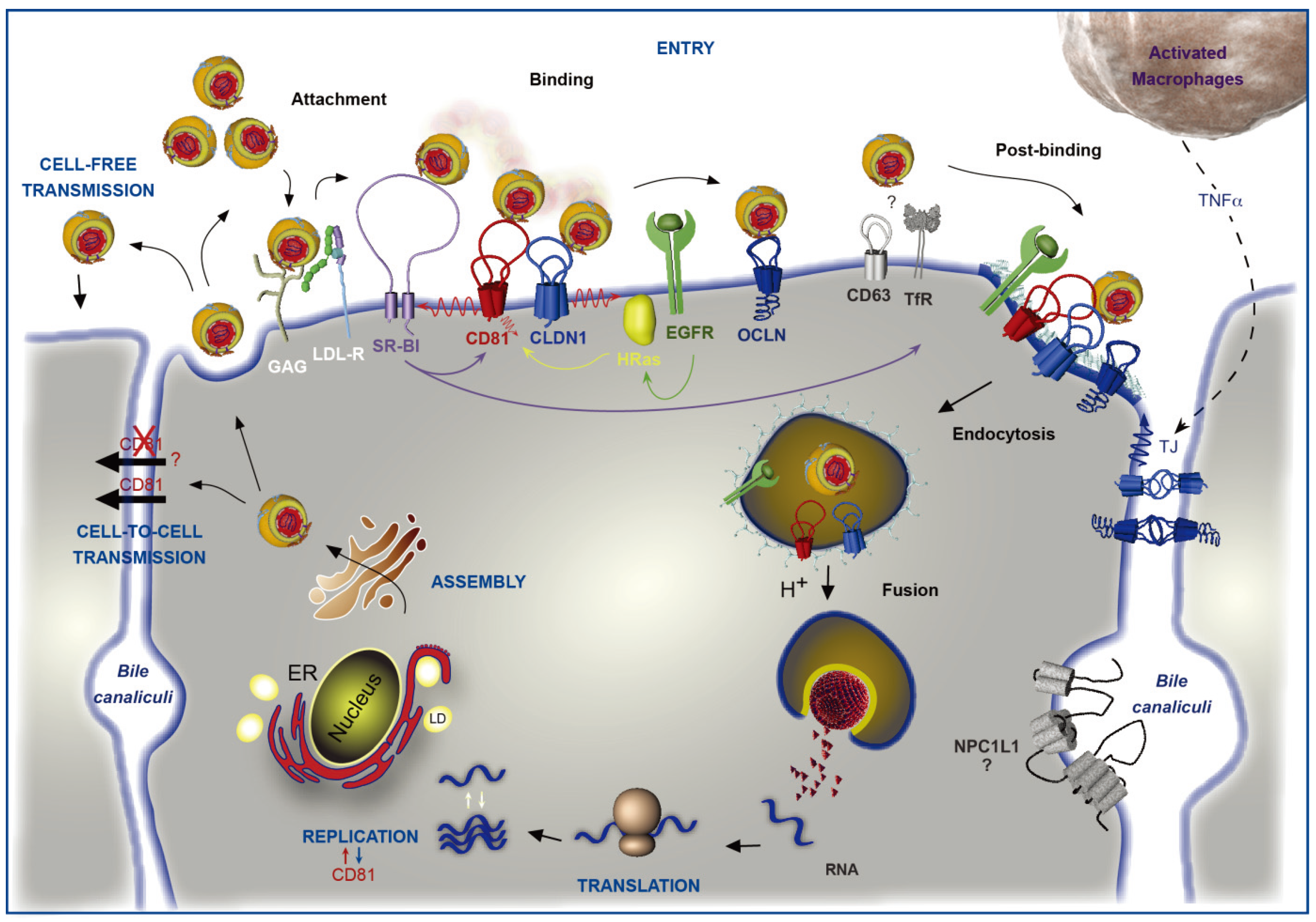
:
1. Introduction

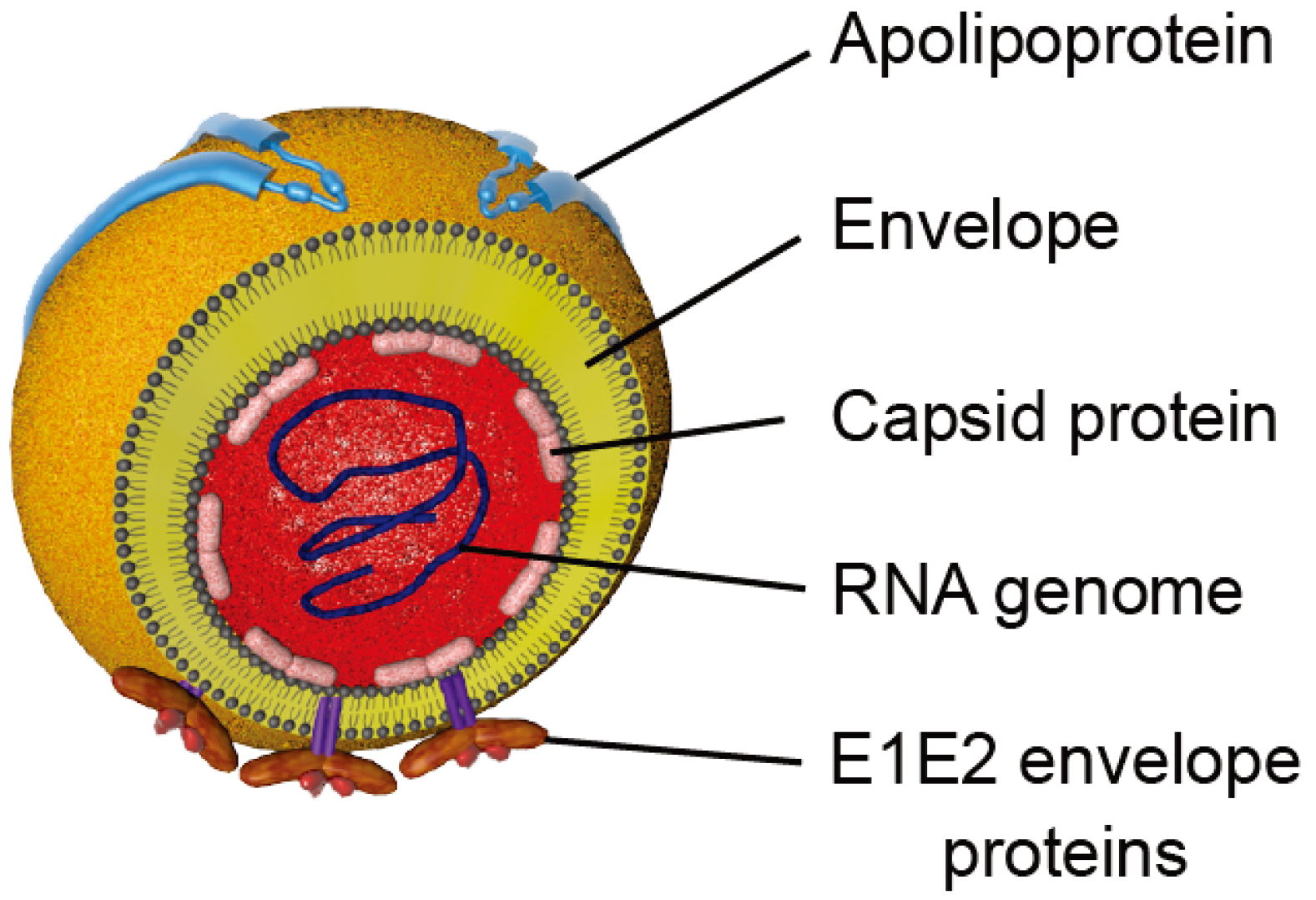
2. HCV Particle and Model Systems to Study the HCV Lifecycle

3. CD81 Plays a Major Role in HCV Entry

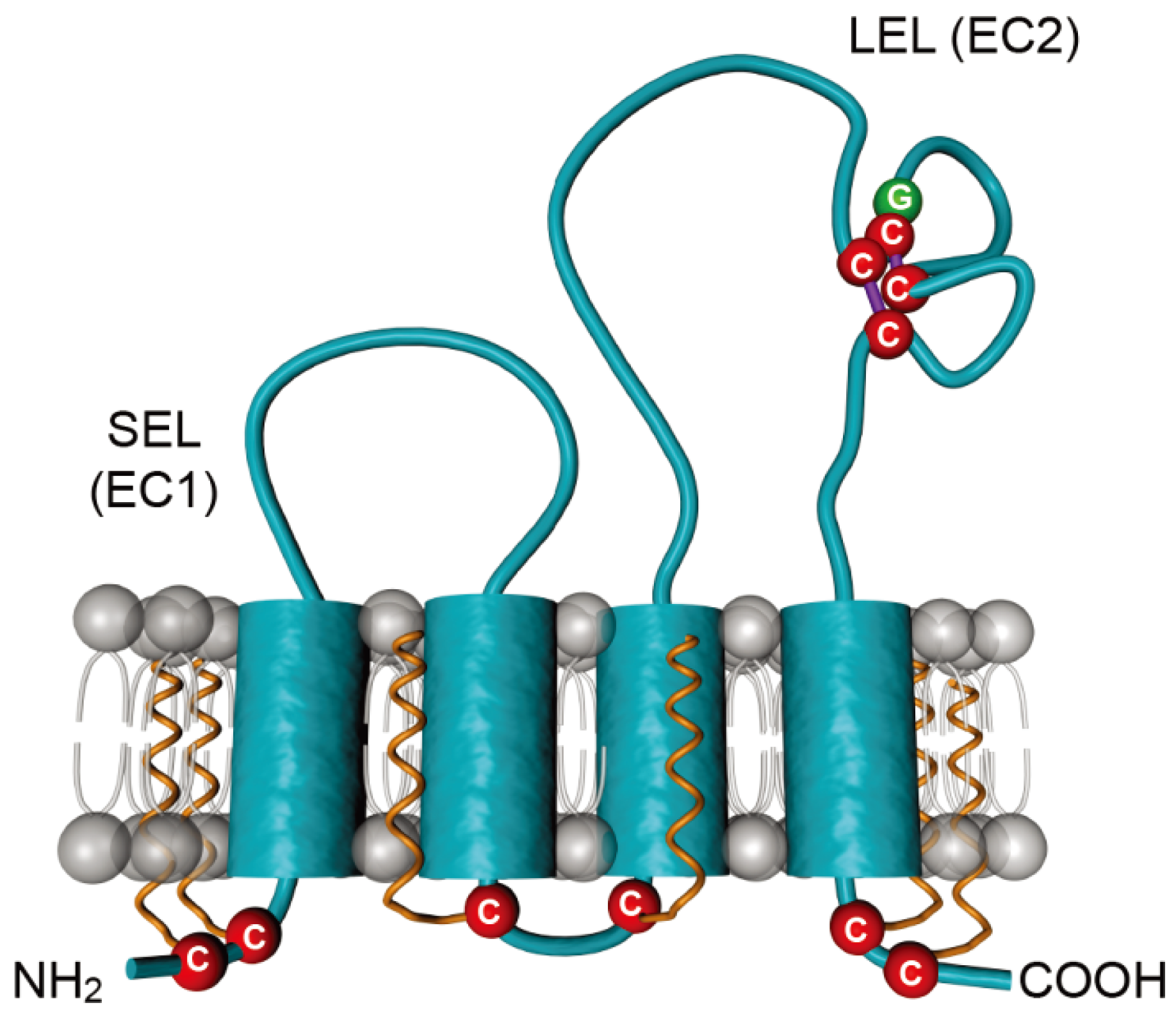
3.1. CD81, a Key Player in HCV Entry
3.2. Determinants in CD81/E2 Interaction
3.2.1. Determinants in CD81
3.2.2. Determinants in E2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E2 residues/regions a | Tools b | Assay | Authors |
|---|---|---|---|
| 480–493/544–551 | E2661 | Blocking antibodies c | [77] |
| 517–535 | E2715 | Blocking antibodies | [105] |
| 474–494/522–551 | E2683 | Modeling | [70] |
| 407–524 | E2660 | Interstrain chimeras | [106] |
| 412–423 | E2660 | Blocking antibodies | [41] |
| 396–407/412–423 | E1E2 | Blocking antibodies | [41] |
| HVRs/613–618 | E2661 | Deletions/Mutagenesis | [68] |
| HVR1 | HCVpp | Deletion | [107] |
| G436WLAGLFY443 | HCVpp | Mutagenesis | [108] |
| 420, 527, 529, 530 and 535 | HCVpp | Blocking antibodies/Mutagenesis | [99,109] |
| 527, 529, 612–619 | HCVpp | Mutagenesis | [110] |
| G451 | HCVcc | Adaptive mutation | [111] |
| N415, 412–423 | HCVcc | Blocking antibodies/Mutagenesis | [112] |
| V388, M405 | HCVcc | Adaptive mutations to mCD81 | [88] |
| DI and DIII domains | E2715 | Modeling | [113] |
| 529–535 | HCVcc | Adaptive mutations to neutralizing antibodies | [97] |
| N415, HVR2 f, IgVR g | HCVcc | Deletion/adaptive mutation | [114] |
| H421 | HCVpp | Mutagenesis | [115] |
| 427, 428, 444 (Front layer) | E2 412–645 | Crystallography Electronic microscopy | [84] |
3.3. CD81 in HCV Species Tropism
3.4. Interplay with Other Entry Factors
3.5. Priming Role of CD81
4. CD81 and Cell-to-Cell Transmission
5. Association of CD81 with EWI Proteins
6. Significance of CD81 Membrane Diffusion in HCV Entry
7. CD81 and HCV Replication
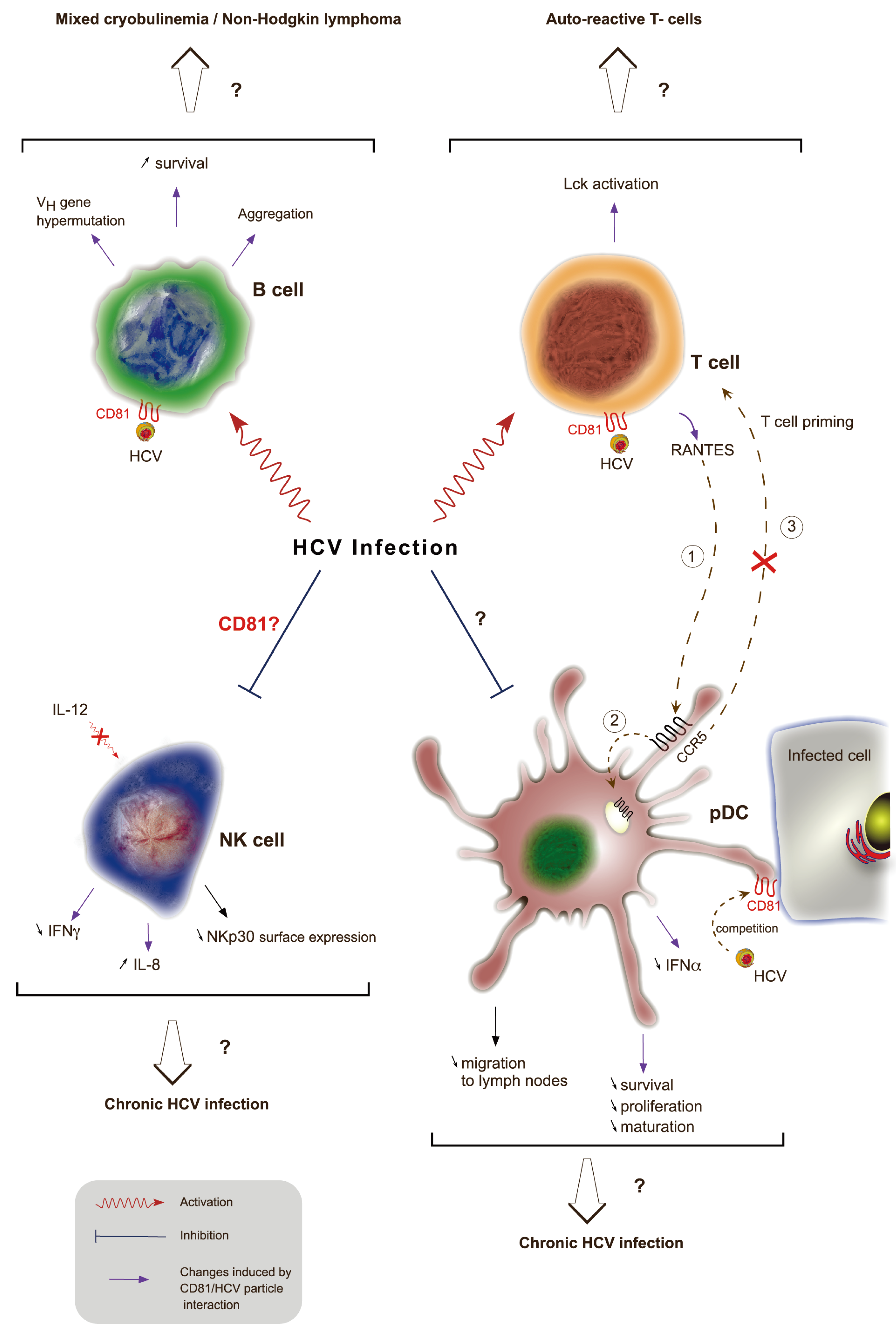
8. Role of CD81 in Immune Response to HCV Infection

9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Oren, R.; Takahashi, S.; Doss, C.; Levy, R.; Levy, S. TAPA-1, the target of an antiproliferative antibody, defines a new family of transmembrane proteins. Mol. Cell. Biol. 1990, 10, 4007–4015. [Google Scholar]
- Van Spriel, A.B. Tetraspanins in the humoral immune response. Biochem. Soc. Trans. 2011, 39, 512–517. [Google Scholar]
- Jones, E.L.; Demaria, M.C.; Wright, M.D. Tetraspanins in cellular immunity. Biochem. Soc. Trans. 2011, 39, 506–511. [Google Scholar] [CrossRef]
- Levy, S.; Todd, S.C.; Maecker, H.T. CD81 (TAPA-1): A molecule involved in signal transduction and cell adhesion in the immune system. Annu. Rev. Immunol. 1998, 16, 89–109. [Google Scholar] [CrossRef]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar]
- Monk, P.N.; Partridge, L.J. Tetraspanins: Gateways for infection. Infect. Disord. Drug Targets 2012, 12, 4–17. [Google Scholar] [CrossRef]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29, 74–81. [Google Scholar] [CrossRef]
- Lemon, S.M.; Walker, C.; Alter, M.J.; Yi, M. Hepatitis C Virus. In Fields Virology; Knipe, D.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 1, pp. 1253–1304. [Google Scholar]
- Manns, M.P.; Foster, G.R.; Rockstroh, J.K.; Zeuzem, S.; Zoulim, F.; Houghton, M. The way forward in HCV treatment —Finding the right path. Nat. Rev. Drug Discov. 2007, 6, 991–1000. [Google Scholar] [CrossRef]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef]
- Zeuzem, S.; Andreone, P.; Pol, S.; Lawitz, E.; Diago, M.; Roberts, S.; Focaccia, R.; Younossi, Z.; Foster, G.R.; Horban, A.; et al. Telaprevir for Retreatment of HCV Infection. N. Engl. J. Med. 2011, 364, 2417–2428. [Google Scholar] [CrossRef]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.-X. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar]
- Molina, S.; Castet, V.; Pichard-Garcia, L.; Wychowski, C.; Meurs, E.; Pascussi, J.-M.; Sureau, C.; Fabre, J.-M.; Sacunha, A.; Larrey, D.; et al. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. J. Virol. 2008, 82, 569–574. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; De Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.-B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar]
- Sainz, B.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; Uprichard, S.L. Identification of the Niemann-Pick C1–like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar]
- Park, J.H.; Park, S.; Yang, J.-S.; Kwon, O.S.; Kim, S.; Jang, S.K. Discovery of cellular proteins required for the early steps of HCV infection using integrative genomics. PLoS One 2013, 8, e60333. [Google Scholar]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Dragic, T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 2006, 80, 11571–11578. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Popescu, C.-I.; Dubuisson, J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell 2010, 102, 63–74. [Google Scholar] [CrossRef]
- Lohmann, V.; Körner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar]
- Blight, K.J. Efficient initiation of HCV RNA replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef]
- Flint, M.; Dubuisson, J.; Maidens, C.; Harrop, R.; Guile, G.R.; Borrow, P.; Mckeating, J.A. Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J. Virol. 2000, 74, 702–709. [Google Scholar] [CrossRef]
- Michalak, J.P.; Wychowski, C.; Choukhi, A.; Meunier, J.C.; Ung, S.; Rice, C.M.; Dubuisson, J. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 1997, 78, 2299–2306. [Google Scholar]
- Deleersnyder, V.; Pillez, A.; Wychowski, C.; Blight, K.; Xu, J.; Hahn, Y.S.; Rice, C.M.; Dubuisson, J. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 1997, 71, 697–704. [Google Scholar]
- Cocquerel, L.; Quinn, E.R.; Flint, M.; Hadlock, K.G.; Foung, S.K. H.; Levy, S. Recognition of native hepatitis C virus E1E2 heterodimers by a human monoclonal antibody. J. Virol. 2003, 77, 1604–1609. [Google Scholar] [CrossRef]
- Op De Beeck, A.; Voisset, C.; Bartosch, B.; Ciczora, Y.; Cocquerel, L.; Keck, Z.; Foung, S.; Cosset, F.-L.; Dubuisson, J. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 2004, 78, 2994–3002. [Google Scholar] [CrossRef]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef]
- Owsianka, A.; Clayton, R.F.; Loomis-Price, L.D.; Mckeating, J.A.; Patel, A.H. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 2001, 82, 1877–1883. [Google Scholar]
- Bartosch, B.; Dubuisson, J.; Cosset, F.-L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2010, 19, 95–103. [Google Scholar] [CrossRef]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar]
- Kitadokoro, K.; Bordo, D.; Galli, G.; Petracca, R.; Falugi, F.; Abrignani, S.; Grandi, G.; Bolognesi, M. CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO J. 2001, 20, 12–18. [Google Scholar]
- Charrin, S.; Manié, S.; Oualid, M.; Billard, M.; Boucheix, C.; Rubinstein, E. Differential stability of tetraspanin/tetraspanin interactions: Role of palmitoylation. FEBS Lett. 2002, 516, 139–144. [Google Scholar]
- Delandre, C.; Penabaz, T.R.; Passarelli, A.L.; Chapes, S.K.; Clem, R.J. Mutation of juxtamembrane cysteines in the tetraspanin CD81 affects palmitoylation and alters interaction with other proteins at the cell surface. Exp. Cell Res. 2009, 315, 1953–1963. [Google Scholar]
- Reynolds, G.M.; Harris, H.J.; Jennings, A.; Hu, K.; Grove, J.; Lalor, P.F.; Adams, D.H.; Balfe, P.; Hubscher, S.G.; McKeating, J.A. Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 2007, 47, 418–427. [Google Scholar]
- Maecker, H.T.; Levy, S. Normal lymphocyte development but delayed humoral immune response in CD81-null mice. J. Exp. Med. 1997, 185, 1505–1510. [Google Scholar] [CrossRef]
- Miyazaki, T.; Müller, U.; Campbell, K.S. Normal development but differentially altered proliferative responses of lymphocytes in mice lacking CD81. EMBO J. 1997, 16, 4217–4225. [Google Scholar] [CrossRef]
- Tsitsikov, E.N.; Gutierrez-Ramos, J.C.; Geha, R.S. Impaired CD19 expression and signaling, enhanced antibody response to type II T independent antigen and reduction of B-1 cells in CD81-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10844–10849. [Google Scholar]
- Rubinstein, E.; Ziyyat, A.; Prenant, M.; Wrobel, E.; Wolf, J.-P.; Levy, S.; Le Naour, F.; Boucheix, C. Reduced fertility of female mice lacking CD81. Dev. Biol. 2006, 290, 351–358. [Google Scholar] [CrossRef]
- Charrin, S.; Latil, M.; Soave, S.; Polesskaya, A.; Chrétien, F.; Boucheix, C.; Rubinstein, E. Normal muscle regeneration requires tight control of muscle cell fusion by tetraspanins CD9 and CD81. Nat. Comms. 2013, 4, 1674. [Google Scholar] [CrossRef]
- Silvie, O.; Rubinstein, E.; Franetich, J.-F.; Prenant, M.; Belnoue, E.; Rénia, L.; Hannoun, L.; Eling, W.; Levy, S.; Boucheix, C.; et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat. Med. 2003, 9, 93–96. [Google Scholar]
- Yalaoui, S.; Zougbédé, S.; Charrin, S.; Silvie, O.; Arduise, C.; Farhati, K.; Boucheix, C.; Mazier, D.; Rubinstein, E.; Froissard, P. Hepatocyte permissiveness to Plasmodium infection is conveyed by a short and structurally conserved region of the CD81 large extracellular domain. PLoS Pathog. 2008, 4, e1000010. [Google Scholar] [CrossRef]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.-L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef]
- Cormier, E.G.; Tsamis, F.; Kajumo, F.; Durso, R.J.; Gardner, J.P.; Dragic, T. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 7270–7274. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Barth, H.; Baumert, T.; McKeating, J.A.; Chisari, F.V. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 2007, 81, 374–383. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef]
- Lavillette, D.; Tarr, A.W.; Voisset, C.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.; Ball, J.K.; Cosset, F.-L. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef]
- Meuleman, P.; Hesselgesser, J.; Paulson, M.; Vanwolleghem, T.; Desombere, I.; Reiser, H.; Leroux-Roels, G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008, 48, 1761–1768. [Google Scholar] [CrossRef]
- Nakajima, H.; Cocquerel, L.; Kiyokawa, N.; Fujimoto, J.; Levy, S. Kinetics of HCV envelope proteins’ interaction with CD81 large extracellular loop. Biochem. Biophys. Res. Commun. 2005, 328, 1091–1100. [Google Scholar] [CrossRef]
- Mckeating, J.A.; Zhang, L.Q.; Logvinoff, C.; Flint, M.; Zhang, J.; Yu, J.; Butera, D.; Ho, D.D.; Dustin, L.B.; Rice, C.M.; et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 2004, 78, 8496–8505. [Google Scholar] [CrossRef]
- Roccasecca, R.; Ansuini, H.; Vitelli, A.; Meola, A.; Scarselli, E.; Acali, S.; Pezzanera, M.; Ercole, B.B.; McKeating, J.; Yagnik, A.; et al. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 2003, 77, 1856–1867. [Google Scholar] [CrossRef]
- Shaw, M.L.; McLauchlan, J.; Mills, P.R.; Patel, A.H.; McCruden, E.A.B. Characterisation of the differences between hepatitis C virus genotype 3 and 1 glycoproteins. J. Med. Virol. 2003, 70, 361–372. [Google Scholar] [CrossRef]
- Yagnik, A.T.; Lahm, A.; Meola, A.; Roccasecca, R.M.; Ercole, B.B.; Nicosia, A.; Tramontano, A. A model for the hepatitis C virus envelope glycoprotein E2. Proteins 2000, 40, 355–366. [Google Scholar] [CrossRef]
- Gottwein, J.M.; Scheel, T.K. H.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef]
- Akazawa, D.; Date, T.; Morikawa, K.; Murayama, A.; Miyamoto, M.; Kaga, M.; Barth, H.; Baumert, T.-F.; Dubuisson, J.; Wakita, T. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 2007, 81, 5036–5045. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Herrmann, E.; Kallis, S.; Bartenschlager, R.; Pietschmann, T. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J. Virol. 2007, 81, 588–598. [Google Scholar]
- Russell, R.S.; Meunier, J.-C.; Takikawa, S.; Faulk, K.; Engle, R.E.; Bukh, J.; Purcell, R.H.; Emerson, S.U. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2008, 105, 4370–4375. [Google Scholar] [CrossRef]
- Rocha-Perugini, V.; Lavie, M.; Delgrange, D.; Canton, J.; Pillez, A.; Potel, J.; Lecoeur, C.; Rubinstein, E.; Dubuisson, J.; Wychowski, C.; et al. The association of CD81 with tetraspanin-enriched microdomains is not essential for hepatitis C virus entry. BMC Microbiol. 2009, 9, 111. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Dixit, N.M. Mathematical model of viral kinetics in vitro estimates the number of E2-CD81 complexes necessary for hepatitis C virus entry. PLoS Comput. Biol. 2011, 7, e1002307. [Google Scholar] [CrossRef]
- Flint, M.; Maidens, C.; Loomis-Price, L.D.; Shotton, C.; Dubuisson, J.; Monk, P.; Higginbottom, A.; Levy, S.; Mckeating, J.A. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 1999, 73, 6235–6244. [Google Scholar]
- Flint, M.; Thomas, J.M.; Maidens, C.M.; Shotton, C.; Levy, S.; Barclay, W.S.; Mckeating, J.A. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol. 1999, 73, 6782–6790. [Google Scholar]
- Masciopinto, F.; Campagnoli, S.; Abrignani, S.; Uematsu, Y.; Pileri, P. The small extracellular loop of CD81 is necessary for optimal surface expression of the large loop, a putative HCV receptor. Virus Res. 2001, 80, 1–10. [Google Scholar] [CrossRef]
- Montpellier, C.; Tews, B.A.; Poitrimole, J.; Rocha-Perugini, V.; D'Arienzo, V.; Potel, J.; Zhang, X.A.; Rubinstein, E.; Dubuisson, J.; Cocquerel, L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J. Biol. Chem. 2011, 286, 13954–13965. [Google Scholar] [CrossRef]
- Higginbottom, A.; Quinn, E.R.; Kuo, C.C.; Flint, M.; Wilson, L.H.; Bianchi, E.; Nicosia, A.; Monk, P.N.; Mckeating, J.A.; Levy, S. Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J. Virol. 2000, 74, 3642–3649. [Google Scholar] [CrossRef]
- Petracca, R.; Falugi, F.; Galli, G.; Norais, N.; Rosa, D.; Campagnoli, S.; Burgio, V.; Di Stasio, E.; Giardina, B.; Houghton, M.; et al. Structure-function analysis of hepatitis C virus envelope-CD81 binding. J. Virol. 2000, 74, 4824–4830. [Google Scholar] [CrossRef]
- Drummer, H.E.; Wilson, K.A.; Poumbourios, P. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J. Virol. 2002, 76, 11143–11147. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C Virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar]
- Rajesh, S.; Sridhar, P.; Tews, B.A.; Feneant, L.; Cocquerel, L.; Ward, D.G.; Berditchevski, F.; Overduin, M. Structural basis of ligand interactions of the large extracellular domain of tetraspanin CD81. J. Virol. 2012, 86, 9606–9616. [Google Scholar] [CrossRef]
- Drummer, H.E.; Wilson, K.A.; Poumbourios, P. Determinants of CD81 dimerization and interaction with hepatitis C virus glycoprotein E2. Biochem. Biophys. Res. Commun. 2005, 328, 251–257. [Google Scholar] [CrossRef]
- Flint, M.; von Hahn, T.; Zhang, J.; Farquhar, M.; Jones, C.T.; Balfe, P.; Rice, C.M.; McKeating, J.A. Diverse CD81 proteins support hepatitis C virus infection. J. Virol. 2006, 80, 11331–11342. [Google Scholar] [CrossRef]
- Bitzegeio, J.; Bankwitz, D.; Hueging, K.; Haid, S.; Brohm, C.; Zeisel, M.-B.; Herrmann, E.; Iken, M.; Ott, M.; Baumert, T.-F.; et al. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog. 2010, 6, e1000978. [Google Scholar] [CrossRef]
- Krieger, S.E.; Zeisel, M.-B.; Davis, C.; Thumann, C.; Harris, H.J.; Schnober, E.K.; Mee, C.; Soulier, E.; Royer, C.; Lambotin, M.; et al. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 2010, 51, 1144–1157. [Google Scholar] [CrossRef]
- Deest, M.; Westhaus, S.; Steinmann, E.; Manns, M.P.; von Hahn, T.; Ciesek, S. Impact of single nucleotide polymorphisms in the essential HCV entry factor CD81 on HCV infectivity and neutralization. Antivir. Res. 2014, 101, 37–44. [Google Scholar] [CrossRef]
- Houldsworth, A.; Metzner, M.M.; Demaine, A.; Hodgkinson, A.; Kaminski, E.; Cramp, M. CD81 sequence and susceptibility to hepatitis C infection. J. Med. Virol. 2014, 86, 162–168. [Google Scholar] [CrossRef]
- Wang, Y.; Keck, Z.-Y.; Foung, S.K. H. Neutralizing antibody response to hepatitis C virus. Viruses 2011, 3, 2127–2145. [Google Scholar] [CrossRef]
- Allander, T.; Forns, X.; Emerson, S.U.; Purcell, R.H.; Bukh, J. Hepatitis C virus envelope protein E2 binds to CD81 of tamarins. Virology 2000, 277, 358–367. [Google Scholar] [CrossRef]
- Keck, Z.-Y.; Op De Beeck, A.; Hadlock, K.G.; Xia, J.; Li, T.-K.; Dubuisson, J.; Foung, S.K.H. Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. J. Virol. 2004, 78, 9224–9232. [Google Scholar] [CrossRef]
- Hadlock, K.G.; Lanford, R.E.; Perkins, S.; Rowe, J.; Yang, Q.; Levy, S.; Pileri, P.; Abrignani, S.; Foung, S.K. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 2000, 74, 10407–10416. [Google Scholar] [CrossRef]
- Keck, Z.-Y.; Li, T.-K.; Xia, J.; Gal-Tanamy, M.; Olson, O.; Li, S.H.; Patel, A.H.; Ball, J.K.; Lemon, S.M.; Foung, S.K.H. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 2008, 82, 6061–6066. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Saha, A.; Xia, J.; Wang, Y.; Lau, P.; Krey, T.; Rey, F.A.; Foung, K.H. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J. Virol. 2011, 85, 10451. [Google Scholar]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.-B.; Baumert, T.-F.; Keck, Z.-Y.; Foung, S.K.H.; et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Timms, J.M.; Tarr, A.W.; Brown, R.J.P.; Hickling, T.P.; Szwejk, A.; Bienkowska-Szewczyk, K.; Thomson, B.J.; Patel, A.H.; Ball, J.K. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 2006, 80, 8695–8704. [Google Scholar] [CrossRef]
- Tarr, A.W.; Owsianka, A.M.; Timms, J.M.; McClure, C.P.; Brown, R.J.P.; Hickling, T.P.; Pietschmann, T.; Bartenschlager, R.; Patel, A.H.; Ball, J.K. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 2006, 43, 592–601. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Robbins, J.B.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A.; Law, M. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. USA 2012, 109, 9499–9504. [Google Scholar]
- Kong, L.; Giang, E.; Nieusma, T.; Robbins, J.B.; Deller, M.C.; Stanfield, R.L.; Wilson, I.A.; Law, M. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J. Virol. 2012, 86, 13085–13088. [Google Scholar] [CrossRef]
- Potter, J.A.; Owsianka, A.M.; Jeffery, N.; Matthews, D.J.; Keck, Z.Y.; Lau, P.; Foung, S.K.H.; Taylor, G.L.; Patel, A.H. Toward a hepatitis C virus vaccine: The structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J. Virol. 2012, 86, 12923–12932. [Google Scholar] [CrossRef]
- Krey, T.; Meola, A.; Keck, Z.-Y.; Damier-Piolle, L.; Foung, S.K.H.; Rey, F.A. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013, 9, e1003364. [Google Scholar] [CrossRef]
- Forns, X.; Allander, T.; Rohwer-Nutter, P.; Bukh, J. Characterization of modified hepatitis C virus E2 proteins expressed on the cell surface. Virology 2000, 274, 75–85. [Google Scholar] [CrossRef]
- Patel, A.H.; Wood, J.; Penin, F.; Dubuisson, J.; Mckeating, J.A. Construction and characterization of chimeric hepatitis C virus E2 glycoproteins: Analysis of regions critical for glycoprotein aggregation and CD81 binding. J. Gen. Virol. 2000, 81, 2873–2883. [Google Scholar]
- Callens, N.; Ciczora, Y.; Bartosch, B.; Vu-Dac, N.; Cosset, F.-L.; Pawlotsky, J.-M.; Penin, F.; Dubuisson, J. Basic residues in hypervariable region 1 of hepatitis C virus envelope glycoprotein e2 contribute to virus entry. J. Virol. 2005, 79, 15331–15341. [Google Scholar] [CrossRef]
- Drummer, H.E.; Boo, I.; Maerz, A.L.; Poumbourios, P. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J. Virol. 2006, 80, 7844–7853. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Tarr, A.W.; Keck, Z.-Y.; Li, T.-K.; Witteveldt, J.; Adair, R.; Foung, S.K.H.; Ball, J.K.; Patel, A.H. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2008, 89, 653–659. [Google Scholar] [CrossRef]
- Rothwangl, K.B.; Manicassamy, B.; Uprichard, S.L.; Rong, L. Dissecting the role of putative CD81 binding regions of E2 in mediating HCV entry: Putative CD81 binding region 1 is not involved in CD81 binding. Virol. J. 2008, 5, 46. [Google Scholar] [CrossRef]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar]
- Dhillon, S.; Witteveldt, J.; Gatherer, D.; Owsianka, A.M.; Zeisel, M.-B.; Zahid, M.N.; Rychłowska, M.; Foung, S.K.H.; Baumert, T.-F.; Angus, A.G.N.; et al. Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J. Virol. 2010, 84, 5494–5507. [Google Scholar] [CrossRef]
- Krey, T.; d'Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; et al. The Disulfide Bonds in Glycoprotein E2 of Hepatitis C Virus Reveal the Tertiary Organization of the Molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, K.; Gouklani, H.; Boo, I.; Poumbourios, P.; Drummer, H.E. The variable regions of hepatitis C virus glycoprotein E2 have an essential structural role in glycoprotein assembly and virion infectivity. J. Gen. Virol. 2011, 92, 112–121. [Google Scholar] [CrossRef]
- Boo, I.; Tewierek, K.; Douam, F.; Lavillette, D.; Poumbourios, P.; Drummer, H. Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidines of HCV glycoprotein E1 and E2. Biochem. J. 2012, 443, 85–94. [Google Scholar]
- Falkowska, E.; Kajumo, F.; Garcia, E.; Reinus, J.; Dragic, T. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 2007, 81, 8072–8079. [Google Scholar] [CrossRef]
- Goffard, A.; Callens, N.; Bartosch, B.; Wychowski, C.; Cosset, F.-L.; Montpellier, C.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 2005, 79, 8400–8409. [Google Scholar]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.-I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.-Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef]
- Lavie, M.L.; Dubuisson, J. HCV glycoprotein assembly of a functional E1-E2 heterodimer. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; pp. 1–30. [Google Scholar]
- Wahid, A.; Helle, F.; Descamps, V.; Duverlie, G.; Penin, F.; Dubuisson, J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J. Virol. 2013, 87, 1605–1617. [Google Scholar]
- Fraser, J.; Boo, I.; Poumbourios, P.; Drummer, H.E. Hepatitis C Virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J. Biol. Chem. 2011, 286, 31984. [Google Scholar]
- Masciopinto, F.; Freer, G.; Burgio, V.L.; Levy, S.; Galli-Stampino, L.; Bendinelli, M.; Houghton, M.; Abrignani, S.; Uematsu, Y. Expression of human CD81 in transgenic mice does not confer susceptibility to hepatitis C virus infection. Virology 2002, 304, 187–196. [Google Scholar] [CrossRef]
- Meola, A.; Sbardellati, A.; Bruni Ercole, B.; Cerretani, M.; Pezzanera, M.; Ceccacci, A.; Vitelli, A.; Levy, S.; Nicosia, A.; Traboni, C.; et al. Binding of hepatitis C virus E2 glycoprotein to CD81 does not correlate with species permissiveness to infection. J. Virol. 2000, 74, 5933–5938. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, Z.-Y.; Klumpp, B.; Wolff-Vorbeck, G.; Barth, H.; Levy, S.; von Weizsacker, F.; Blum, H.E.; Baumert, T.-F. Primary hepatocytes of Tupaia belangeri as a potential model for hepatitis C virus infection. J. Clin. Invest. 2002, 109, 221–232. [Google Scholar] [CrossRef]
- Xie, Z.C.; Riezu-Boj, J.I.; Lasarte, J.J.; Guillen, J.; Su, J.H.; Civeira, M.P.; Prieto, J. Transmission of hepatitis C virus infection to tree shrews. Virology 1998, 244, 513–520. [Google Scholar] [CrossRef]
- Xu, X.; Chen, H.; Cao, X.; Ben, K. Efficient infection of tree shrew (Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma. J. Gen. Virol. 2007, 88, 2504–2512. [Google Scholar] [CrossRef]
- Tian, Z.-F.; Shen, H.; Fu, X.-H.; Chen, Y.-C.; Blum, H.E.; Baumert, T.-F.; Zhao, X.-P. Interaction of hepatitis C virus envelope glycoprotein E2 with the large extracellular loop of tupaia CD81. World J. Gastroenterol. 2009, 15, 240–244. [Google Scholar]
- Tong, Y.; Zhu, Y.; Xia, X.; Liu, Y.; Feng, Y.; Hua, X.; Chen, Z.; Ding, H.; Gao, L.; Wang, Y.; et al. Tupaia CD81, SR-BI, claudin-1, and occludin support hepatitis C virus infection. J. Virol. 2011, 85, 2793–2802. [Google Scholar] [CrossRef]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef]
- Dorner, M.; Horwitz, J.A.; Donovan, B.M.; Labitt, R.N.; Budell, W.C.; Friling, T.; Vogt, A.; Catanese, M.T.; Satoh, T.; Kawai, T.; et al. Completion of the entire hepatitis c virus life cycle in genetically humanized mice. Nature 2013, 501, 237–241. [Google Scholar] [CrossRef]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Catanese, M.T.; Graziani, R.; von Hahn, T.; Moreau, M.; Huby, T.; Paonessa, G.; Santini, C.; Luzzago, A.; Rice, C.M.; Cortese, R.; et al. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 2007, 81, 8063–8071. [Google Scholar] [CrossRef]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; et al. Role of scavenger receptor class B type I in hepatitis C virus entry: Kinetics and molecular determinants. J. Virol. 2010, 84, 34–43. [Google Scholar] [CrossRef]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guérin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.-L.; et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef]
- Grove, J.; Huby, T.; Stamataki, Z.; Vanwolleghem, T.; Meuleman, P.; Farquhar, M.; Schwarz, A.; Moreau, M.; Owen, J.S.; Leroux-Roels, G.; et al. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 2007, 81, 3162–3169. [Google Scholar] [CrossRef]
- Zeisel, M.-B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.-L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef]
- Schwarz, A.K.; Grove, J.; Hu, K.; Mee, C.J.; Balfe, P.; McKeating, J.A. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J. Virol. 2009, 83, 12407–12414. [Google Scholar] [CrossRef]
- Zahid, M.N.; Turek, M.; Xiao, F.; Dao Thi, V.L.; Guérin, M.; Fofana, I.; Bachellier, P.; Thompson, J.; Delang, L.; Neyts, J.; et al. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 2012, 57, 492–504. [Google Scholar] [CrossRef]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insightswith implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Dreux, M.; Cosset, F.-L. Scavenger receptor class B type I and the hypervariable region-1 of hepatitis C virus in cell entry and neutralisation. Expert Rev. Mol. Med. 2011, 13, e13. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.-L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef]
- Zheng, A.; Yuan, F.; Li, Y.; Zhu, F.; Hou, P.; Li, J.; Song, X.; Ding, M.; Deng, H. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 2007, 81, 12465–12471. [Google Scholar] [CrossRef]
- Douam, F.; Thi, V.L.D.; Maurin, G.; Fresquet, J.; Mompelat, D.; Zeisel, M.-B.; Baumert, T.-F.; Cosset, F.-L.; Lavillette, D. A critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2013. [Google Scholar] [CrossRef]
- Fofana, I.; Krieger, S.E.; Grunert, F.; Glauben, S.; Xiao, F.; Fafi-Kremer, S.; Soulier, E.; Royer, C.; Thumann, C.; Mee, C.J.; et al. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 2010, 139, 953–64–964.e1–4. [Google Scholar] [CrossRef]
- Benedicto, I.; Molina-Jiménez, F.; Bartosch, B.; Cosset, F.-L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernández, A.; Aldabe, R.; López-Cabrera, M.; et al. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef]
- Yang, W.; Qiu, C.; Biswas, N.; Jin, J.; Watkins, S.C.; Montelaro, R.C.; Coyne, C.B.; Wang, T. Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J. Biol. Chem. 2008, 283, 8643–8653. [Google Scholar]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar]
- Harris, H.J.; Davis, C.; Mullins, J.G.L.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar]
- Mee, C.J.; Harris, H.J.; Farquhar, M.J.; Wilson, G.; Reynolds, G.; Davis, C.; van Ijzendoorn, S.C.D.; Balfe, P.; McKeating, J.A. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J. Virol. 2009, 83, 6211–6221. [Google Scholar] [CrossRef]
- Davis, C.; Harris, H.J.; Hu, K.; Drummer, H.E.; McKeating, J.A.; Mullins, J.G.L.; Balfe, P. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell. Microbiol. 2012, 14, 1892–1903. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Dell'Angelica, E.C. Molecular bases for the recognition of tyrosine-based sorting signals. J. Cell Biol. 1999, 145, 923–926. [Google Scholar] [CrossRef]
- Haucke, V.; Krauss, M. Tyrosine-based endocytic motifs stimulate oligomerization of AP-2 adaptor complexes. Eur. J. Cell Biol. 2002, 81, 647–653. [Google Scholar] [CrossRef]
- Utech, M.; Mennigen, R.; Bruewer, M. Endocytosis and recycling of tight junction proteins in inflammation. J. Biomed. Biotechnol. 2010, 2010, 484987. [Google Scholar]
- Yu, D.; Turner, J.R. Stimulus-induced reorganization of tight junction structure: The role of membrane traffic. Biochim. Biophys. Acta 2008, 1778, 709–716. [Google Scholar]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; Mckeating, J.A. Hepatitis C virus induces CD81 and Claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef]
- Coyne, C.B.; Bergelson, J.M. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 2006, 124, 119–131. [Google Scholar]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008, 82, 8316–8329. [Google Scholar] [CrossRef]
- Reynolds, G.M.; Harris, H.J.; Jennings, A.; Hu, K.; Grove, J.; Lalor, P.F.; Adams, D.H.; Balfe, P.; Hübscher, S.G.; McKeating, J.A. Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 2008, 47, 418–427. [Google Scholar]
- Heiskala, M.; Peterson, P.A.; Yang, Y. The roles of claudin superfamily proteins in paracellular transport. Traffic 2001, 2, 93–98. [Google Scholar]
- Mee, C.J.; Grove, J.; Harris, H.J.; Hu, K.; Balfe, P.; McKeating, J.A. Effect of cell polarization on hepatitis C virus entry. J. Virol. 2008, 82, 461–470. [Google Scholar] [CrossRef]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef]
- Sourisseau, M.; Michta, M.L.; Zony, C.; Israelow, B.; Hopcraft, S.E.; Narbus, C.M.; Parra Martín, A.; Evans, M.J. Temporal analysis of Hepatitis C Virus cell entry with occludin directed blocking antibodies. PLoS Pathog. 2013, 9, e1003244. [Google Scholar] [CrossRef]
- Liu, S.; Kuo, W.; Yang, W.; Liu, W.; Gibson, G.A.; Dorko, K.; Watkins, S.C.; Strom, S.C.; Wang, T. The second extracellular loop dictates Occludin-mediated HCV entry. Virology 2010, 407, 160–170. [Google Scholar] [CrossRef]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.-L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar]
- Chang, M.; Williams, O.; Mittler, J.; Quintanilla, A.; Carithers, R.L.; Perkins, J.; Corey, L.; Gretch, D.R. Dynamics of hepatitis C virus replication in human liver. Am. J. Pathol. 2003, 163, 433–444. [Google Scholar]
- Wieland, S.; Makowska, Z.; Campana, B.; Calabrese, D.; Dill, M.T.; Chung, J.; Chisari, F.V.; Heim, M.H. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2013. [Google Scholar] [CrossRef]
- Brimacombe, C.L.; Grove, J.; Meredith, L.W.; Hu, K.; Syder, A.J.; Flores, M.V.; Timpe, J.M.; Krieger, S.E.; Baumert, T.-F.; Tellinghuisen, T.L.; et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 2011, 85, 596–605. [Google Scholar] [CrossRef]
- Ciesek, S.; Westhaus, S.; Wicht, M.; Wappler, I.; Henschen, S.; Sarrazin, C.; Hamdi, N.; Abdelaziz, A.I.; Strassburg, C.P.; Wedemeyer, H.; et al. Impact of intra- and interspecies variation of occludin on its function as coreceptor for authentic hepatitis C virus particles. J. Virol. 2011, 85, 7613–7621. [Google Scholar] [CrossRef]
- Meredith, L.W.; Harris, H.J.; Wilson, G.K.; Fletcher, N.F.; Balfe, P.; McKeating, J.A. Early infection events highlight the limited transmissibility of hepatitis C virus in vitro. J. Hepatol. 2013, 58, 1074–1080. [Google Scholar] [CrossRef]
- Catanese, M.T.; Loureiro, J.; Jones, C.T.; Dorner, M.; von Hahn, T.; Rice, C.M. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J. Virol. 2013, 87, 8282–8293. [Google Scholar]
- Valli, M.B.; Crema, A.; Lanzilli, G.; Serafino, A.; Bertolini, L.; Ravagnan, G.; Ponzetto, A.; Menzo, S.; Clementi, M.; Carloni, G. Molecular and cellular determinants of cell-to-cell transmission of HCV in vitro. J. Med. Virol. 2007, 79, 1491–1499. [Google Scholar] [CrossRef]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar]
- Witteveldt, J.; Evans, M.J.; Bitzegeio, J.; Koutsoudakis, G.; Owsianka, A.M.; Angus, A.G.N.; Keck, Z.-Y.; Foung, S.K.H.; Pietschmann, T.; Rice, C.M.; et al. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 2009, 90, 48–58. [Google Scholar] [CrossRef]
- Jones, C.T.; Catanese, M.T.; Law, L.M.J.; Khetani, S.R.; Syder, A.J.; Ploss, A.; Oh, T.S.; Schoggins, J.W.; MacDonald, M.R.; Bhatia, S.N.; et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 2010, 28, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Potel, J.; Rassam, P.; Montpellier, C.; Kaestner, L.; Werkmeister, E.; Tews, B.A.; Couturier, C.; Popescu, C.-I.; Baumert, T.-F.; Rubinstein, E.; et al. EWI-2wint promotes CD81 clustering that abrogates hepatitis C virus entry. Cell. Microbiol. 2013, 15, 1234–1252. [Google Scholar]
- Sattentau, Q.J. Cell-to-cell spread of retroviruses. Viruses 2010, 2, 1306–1321. [Google Scholar] [CrossRef]
- Perez-Hernandez, D.; Gutierrez-Vazquez, C.; Jorge, I.; Lopez-Martin, S.; Ursa, A.; Sánchez Madrid, F.; Vazquez, J.; Yáñez-Mó, M. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J. Biol. Chem. 2013, 288, 11649–11661. [Google Scholar] [CrossRef]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef]
- Charrin, S.; Le Naour, F.; Silvie, O.; Milhiet, P.-E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: Tetraspanins spin their web. Biochem. J. 2009, 420, 133–154. [Google Scholar]
- Bradbury, L.E.; Kansas, G.S.; Levy, S.; Evans, R.L.; Tedder, T.F. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J. Immunol. 1992, 149, 2841–2850. [Google Scholar]
- Imai, T.; Yoshie, O. C33 antigen and M38 antigen recognized by monoclonal antibodies inhibitory to syncytium formation by human T cell leukemia virus type 1 are both members of the transmembrane 4 superfamily and associate with each other and with CD4 or CD8 in T cells. J. Immunol. 1993, 151, 6470–6481. [Google Scholar]
- Serru, V.; Le Naour, F.; Billard, M.; Azorsa, D.O.; Lanza, F.; Boucheix, C.; Rubinstein, E. Selective tetraspan-integrin complexes (CD81/alpha4beta1, CD151/alpha3beta1, CD151/alpha6beta1) under conditions disrupting tetraspan interactions. Biochem. J. 1999, 340, 103–111. [Google Scholar]
- Rubinstein, E.; Le Naour, F.; Lagaudrière-Gesbert, C.; Billard, M.; Conjeaud, H.; Boucheix, C. CD9, CD63, CD81, and CD82 are components of a surface tetraspan network connected to HLA-DR and VLA integrins. Eur. J. Immunol. 1996, 26, 2657–2665. [Google Scholar] [CrossRef]
- Charrin, S.; Le Naour, F.; Labas, V.; Billard, M.; Le Caer, J.-P.; Emile, J.-F.; Petit, M.-A.; Boucheix, C.; Rubinstein, E. EWI-2 is a new component of the tetraspanin web in hepatocytes and lymphoid cells. Biochem. J. 2003, 373, 409–421. [Google Scholar] [CrossRef]
- Charrin, S.; Le Naour, F.; Oualid, M.; Billard, M.; Faure, G.; Hanash, S.M.; Boucheix, C.; Rubinstein, E. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J. Biol. Chem. 2001, 276, 14329–14337. [Google Scholar]
- Clark, K.L.; Zeng, Z.; Langford, A.L.; Bowen, S.M.; Todd, S.C. PGRL is a major CD81-associated protein on lymphocytes and distinguishes a new family of cell surface proteins. J. Immunol. 2001, 167, 5115–5121. [Google Scholar]
- Rocha-Perugini, V.; Montpellier, C.; Delgrange, D.; Wychowski, C.; Helle, F.; Pillez, A.; Drobecq, H.; Le Naour, F.; Charrin, S.; Levy, S.; et al. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 2008, 3, e1866. [Google Scholar] [CrossRef]
- Stipp, C.S.; Kolesnikova, T.V.; Hemler, M.E. EWI-2 is a major CD9 and CD81 partner and member of a novel Ig protein subfamily. J. Biol. Chem. 2001, 276, 40545–40554. [Google Scholar] [CrossRef]
- Stipp, C.S.; Orlicky, D.; Hemler, M.E. FPRP, a major, highly stoichiometric, highly specific CD81- and CD9-associated protein. J. Biol. Chem. 2001, 276, 4853–4862. [Google Scholar] [CrossRef]
- Sala-Valdés, M.; Ursa, A.; Charrin, S.; Rubinstein, E.; Hemler, M.E.; Sánchez-Madrid, F.; Yáñez-Mó, M. EWI-2 and EWI-F link the tetraspanin web to the actin cytoskeleton through their direct association with ezrin-radixin-moesin proteins. J. Biol. Chem. 2006, 281, 19665–19675. [Google Scholar] [CrossRef]
- Stipp, C.S.; Kolesnikova, T.V.; Hemler, M.E. EWI-2 regulates alpha3beta1 integrin-dependent cell functions on laminin-5. J. Cell Biol. 2003, 163, 1167–1177. [Google Scholar] [CrossRef]
- Zhang, X.A.; Lane, W.S.; Charrin, S.; Rubinstein, E.; Liu, L. EWI2/PGRL associates with the metastasis suppressor KAI1/CD82 and inhibits the migration of prostate cancer cells. Canc. Res. 2003, 63, 2665–2674. [Google Scholar]
- Charrin, S.; Yalaoui, S.; Bartosch, B.; Cocquerel, L.; Franetich, J.-F.; Boucheix, C.; Mazier, D.; Rubinstein, E.; Silvie, O. The Ig domain protein CD9P-1 down-regulates CD81 ability to support Plasmodium yoelii infection. J. Biol. Chem. 2009, 284, 31572–31578. [Google Scholar] [CrossRef]
- Burckhardt, C.J.; Greber, U.F. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.J.; Clerte, C.; Farquhar, M.J.; Goodall, M.; Hu, K.; Rassam, P.; Dosset, P.; Wilson, G.K.; Balfe, P.; Ijzendoorn, S.C.; et al. Hepatoma polarization limits CD81 and hepatitis C virus dynamics. Cell. Microbiol. 2012, 15, 430–445. [Google Scholar]
- Espenel, C.; Margeat, E.; Dosset, P.; Arduise, C.; Le Grimellec, C.; Royer, C.A.; Boucheix, C.; Rubinstein, E.; Milhiet, P.-E. Single-molecule analysis of CD9 dynamics and partitioning reveals multiple modes of interaction in the tetraspanin web. J. Cell Biol. 2008, 182, 765–776. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Rassam, P.; Margeat, E.; Roy, N.H.; Schneider-Schaulies, J.; Milhiet, P.-E.; Thali, M. HIV-1 Assembly Differentially Alters Dynamics and Partitioning of Tetraspanins and Raft Components. Traffic 2010, 11, 1401–1414. [Google Scholar] [CrossRef]
- Fletcher, N.F.; Sutaria, R.; Jo, J.; Barnes, A.; Blahova, M.; Meredith, L.W.; Cosset, F.-L.; Curbishley, S.M.; Adams, D.H.; Bertoletti, A.; et al. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology 2013. [Google Scholar] [CrossRef]
- Masciopinto, F.; Giovani, C.; Campagnoli, S.; Galli-Stampino, L.; Colombatto, P.; Brunetto, M.; Yen, T.S.B.; Houghton, M.; Pileri, P.; Abrignani, S. Association of hepatitisC virus envelope proteins with exosomes. Eur. J. Immunol. 2004, 34, 2834–2842. [Google Scholar] [CrossRef]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Zhang, B.-H.; Ishii, K.; Liang, T.J. Novel function of CD81 in controlling hepatitis C virus replication. J. Virol. 2010, 84, 3396–3407. [Google Scholar] [CrossRef]
- Ke, P.-Y.; Chen, S.S.-L. Active RNA replication of hepatitis C virus downregulates CD81 expression. PLoS One 2013, 8, e54866. [Google Scholar]
- Tscherne, D.M.; Evans, M.J.; von Hahn, T.; Jones, C.T.; Stamataki, Z.; McKeating, J.A.; Lindenbach, B.D.; Rice, C.M. Superinfection exclusion in cells infected with hepatitis C virus. J. Virol. 2007, 81, 3693–3703. [Google Scholar] [CrossRef]
- Zignego, A.L.; Giannini, C.; Gragnani, L. HCV and Lymphoproliferation. Clin. Dev. Immunol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Agnello, V.; Chung, R.T.; Kaplan, L.M. A role for hepatitis C virus infection in type II cryoglobulinemia. N. Engl. J. Med. 1992, 327, 1490–1495. [Google Scholar] [CrossRef]
- Engels, E.A.; Chatterjee, N.; Cerhan, J.R.; Davis, S.; Cozen, W.; Severson, R.K.; Whitby, D.; Colt, J.S.; Hartge, P. Hepatitis C virus infection and non-hodgkin lymphoma: Results of the NCI-seer multi-center case-control study. Int. J. Canc. 2004, 111, 76–80. [Google Scholar] [CrossRef]
- Sansonno, D.; Carbone, A.; De Re, V.; Dammacco, F. Hepatitis C virus infection, cryoglobulinaemia, and beyond. Rheumatology (Oxford) 2007, 46, 572–578. [Google Scholar]
- Cocquerel, L.; Kuo, C.-C.; Dubuisson, J.; Levy, S. CD81-dependent binding of hepatitis C virus E1E2 heterodimers. J. Virol. 2003, 77, 10677–10683. [Google Scholar] [CrossRef]
- Rosa, D.; Saletti, G.; De Gregorio, E.; Zorat, F.; Comar, C.; D'Oro, U.; Nuti, S.; Houghton, M.; Barnaba, V.; Pozzato, G.; et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 18544–18549. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T. H.; Pavio, N.; Sung, V.M.H.; Lai, M.M. C. Hepatitis C Virus E2-CD81 interaction induces hypermutation of the immunoglobulin gene in B cells. J. Virol. 2005, 79, 8079–8089. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, Y.; Ren, Y.; Tong, Y.; Hua, X.; Zhu, F.; Huang, L.; Liu, Y.; Luo, Y.; Lu, W.; Zhao, P.; Qi, Z. Hepatitis C Virus protects human B lymphocytes from Fas-Mediated apoptosis via E2-CD81 engagement. PLoS One 2011, 6, e18933. [Google Scholar] [CrossRef]
- Ni, J.; Hembrador, E.; Di Bisceglie, A.M.; Jacobson, I.M.; Talal, A.H.; Butera, D.; Rice, C.M.; Chambers, T.J.; Dustin, L.B. Accumulation of B lymphocytes with a naive, resting phenotype in a subset of hepatitis C patients. J. Immunol. 2003, 170, 3429–3439. [Google Scholar]
- Takahashi, S.; Doss, C.; Levy, S.; Levy, R. TAPA-1, the target of an antiproliferative antibody, is associated on the cell surface with the Leu-13 antigen. J. Immunol. 1990, 145, 2207–2213. [Google Scholar]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Kim, H.; Ray, R.; Ray, R.B. ISG56 and IFITM1 Proteins Inhibit Hepatitis C Virus Replication. J. Virol. 2011, 85, 12881–12889. [Google Scholar] [CrossRef]
- Wilkins, C.; Woodward, J.; Lau, D.T. Y.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2012, 57, 461–469. [Google Scholar]
- Tseng, C.T.K.; Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–50. [Google Scholar] [CrossRef]
- Crotta, S.; Stilla, A.; Wack, A.; D'Andrea, A.; Nuti, S.; D'Oro, U.; Mosca, M.; Filliponi, F.; Brunetto, R.M.; Bonino, F.; et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 2002, 195, 35–42. [Google Scholar] [CrossRef]
- Crotta, S.; Ronconi, V.; Ulivieri, C.; Baldari, C.T.; Valiante, N.M.; Valiente, N.M.; Abrignani, S.; Wack, A. Cytoskeleton rearrangement induced by tetraspanin engagement modulates the activation of T and NK cells. Eur. J. Immunol. 2006, 36, 919–929. [Google Scholar] [CrossRef]
- Corado, J.; Toro, F.; Rivera, H.; Bianco, N.E.; Deibis, L.; De Santis, J.B. Impairment of natural killer (NK) cytotoxic activity in hepatitis C virus (HCV) infection. Clin. Exp. Immunol. 1997, 109, 451–457. [Google Scholar]
- Yoon, J.C.; Shiina, M.; Ahlenstiel, G.; Rehermann, B. Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology 2009, 49, 12–21. [Google Scholar] [CrossRef]
- Holder, K.A.; Stapleton, S.N.; Gallant, M.E.; Russell, R.S.; Grant, M.D. Hepatitis C Virus-infected cells downregulate NKp30 and inhibit ex vivo NK cell functions. J. Immunol. 2013, 191, 3308–3318. [Google Scholar] [CrossRef]
- Farag, M.M.S.; Weigand, K.; Encke, J.; Momburg, F. Activation of natural killer cells by hepatitis C virus particles in vitro. Clin. Exp. Immunol. 2011, 165, 352–362. [Google Scholar] [CrossRef]
- Crotta, S.; Brazzoli, M.; Piccioli, D.; Valiante, N.M.; Wack, A. Hepatitis C virions subvert natural killer cell activation to generate a cytokine environment permissive for infection. J. Hepatol. 2010, 52, 183–190. [Google Scholar] [CrossRef]
- Zhang, S.; Saha, B.; Kodys, K.; Szabo, G. IFN-γ production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J. Hepatol. 2013, 59, 442–449. [Google Scholar] [CrossRef]
- Tseng, C. Characterization of liver T-cell receptor γδ+ T cells obtained from individuals chronically infected with hepatitis C virus (HCV): Evidence for these T cells playing a role in the liver pathology associated with HCV infections. Hepatology 2001, 33, 1312–1320. [Google Scholar] [CrossRef]
- Tseng, C.-T.K.; Miskovsky, E.; Klimpel, G.R. Crosslinking CD81 Results in Activation of TCRγδ T Cells. Cell. Immunol. 2001, 207, 19–27. [Google Scholar]
- Wack, A.; Soldaini, E.; Tseng, C.-T.K.; Nuti, S.; Klimpel, G.R.; Abrignani, S. Binding of the hepatitis C virus envelope protein E2 to CD81 provides a co-stimulatory signal for human T cells. Eur. J. Immunol. 2001, 31, 166–175. [Google Scholar]
- Soldaini, E.; Wack, A.; D'Oro, U.; Nuti, S.; Ulivieri, C.; Baldari, C.T.; Abrignani, S. T cell costimulation by the hepatitis C virus envelope protein E2 binding to CD81 is mediated by Lck. Eur. J. Immunol. 2003, 33, 455–464. [Google Scholar]
- Averill, L.; Lee, W.M.; Karandikar, N.J. Differential dysfunction in dendritic cell subsets during chronic HCV infection. Clin. Immunol. 2007, 123, 40–49. [Google Scholar]
- Szabo, G.; Dolganiuc, A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology 2005, 210, 237–247. [Google Scholar] [CrossRef]
- Lai, W.K.; Curbishley, S.M.; Goddard, S.; Alabraba, E.; Shaw, J.; Youster, J.; McKeating, J.; Adams, D.H. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J. Hepatol. 2007, 47, 338–347. [Google Scholar] [CrossRef]
- Shiina, M.; Rehermann, B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology 2007, 47, 385–395. [Google Scholar]
- Liang, H.; Russell, R.S.; Yonkers, N.L.; McDonald, D.; Rodriguez, B.; Harding, C.V.; Anthony, D.D. Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J. Virol. 2009, 83, 5693–5707. [Google Scholar] [CrossRef]
- Longman, R.S.; Talal, A.H.; Jacobson, I.M.; Rice, C.M.; Albert, M.L. Normal functional capacity in circulating myeloid and plasmacytoid dendritic cells in patients with chronic hepatitis C. J. Infect. Dis. 2005, 192, 497–503. [Google Scholar] [CrossRef]
- Piccioli, D.; Tavarini, S.; Nuti, S.; Colombatto, P.; Brunetto, M.; Bonino, F.; Ciccorossi, P.; Zorat, F.; Pozzato, G.; Comar, C.; et al. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J. Hepatol. 2005, 42, 61–67. [Google Scholar]
- Albert, M.L.; Decalf, J.; Pol, S. Plasmacytoid dendritic cells move down on the list of suspects: In search of the immune pathogenesis of chronic hepatitis C. J. Hepatol. 2008, 49, 1069–1078. [Google Scholar] [CrossRef]
- Nattermann, J.; Zimmermann, H.; Iwan, A.; von Lilienfeld-Toal, M.; Leifeld, L.; Nischalke, H.D.; Langhans, B.; Sauerbruch, T.; Spengler, U. Hepatitis C virus E2 and CD81 interaction may be associated with altered trafficking of dendritic cells in chronic hepatitis C. Hepatology 2006, 44, 945–954. [Google Scholar] [CrossRef]
- Zhang, S.; Kodys, K.; Babcock, G.J.; Szabo, G. CD81/CD9 tetraspanins aid plasmacytoid dendritic cells in recognition of hepatitis C virus-infected cells and induction of interferon-alpha. Hepatology 2013, 58, 940–949. [Google Scholar] [CrossRef]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar]
- Tu, Z.; Zhang, P.; Li, H.; Niu, J.; Jin, X.; Su, L. Cellular Immunology. Cell. Immunol. 2013, 284, 98–103. [Google Scholar] [CrossRef]
- Zona, L.; Tawar, R.G.; Zeisel, M.B.; Xiao, F.; Schuster, C.; Lupberger, J.; Baumert, T.F. Tetraspanin-co-receptor associations—Impact for hepatitis C virus entry and antiviral therapies. Viruses 2014. submitted for publication. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fénéant, L.; Levy, S.; Cocquerel, L. CD81 and Hepatitis C Virus (HCV) Infection. Viruses 2014, 6, 535-572. https://doi.org/10.3390/v6020535
Fénéant L, Levy S, Cocquerel L. CD81 and Hepatitis C Virus (HCV) Infection. Viruses. 2014; 6(2):535-572. https://doi.org/10.3390/v6020535
Chicago/Turabian StyleFénéant, Lucie, Shoshana Levy, and Laurence Cocquerel. 2014. "CD81 and Hepatitis C Virus (HCV) Infection" Viruses 6, no. 2: 535-572. https://doi.org/10.3390/v6020535
APA StyleFénéant, L., Levy, S., & Cocquerel, L. (2014). CD81 and Hepatitis C Virus (HCV) Infection. Viruses, 6(2), 535-572. https://doi.org/10.3390/v6020535