Quantifying Susceptibility of CD4+ Stem Memory T-Cells to Infection by Laboratory Adapted and Clinical HIV-1 Strains

Abstract
:1. Introduction
2. Results and Discussion
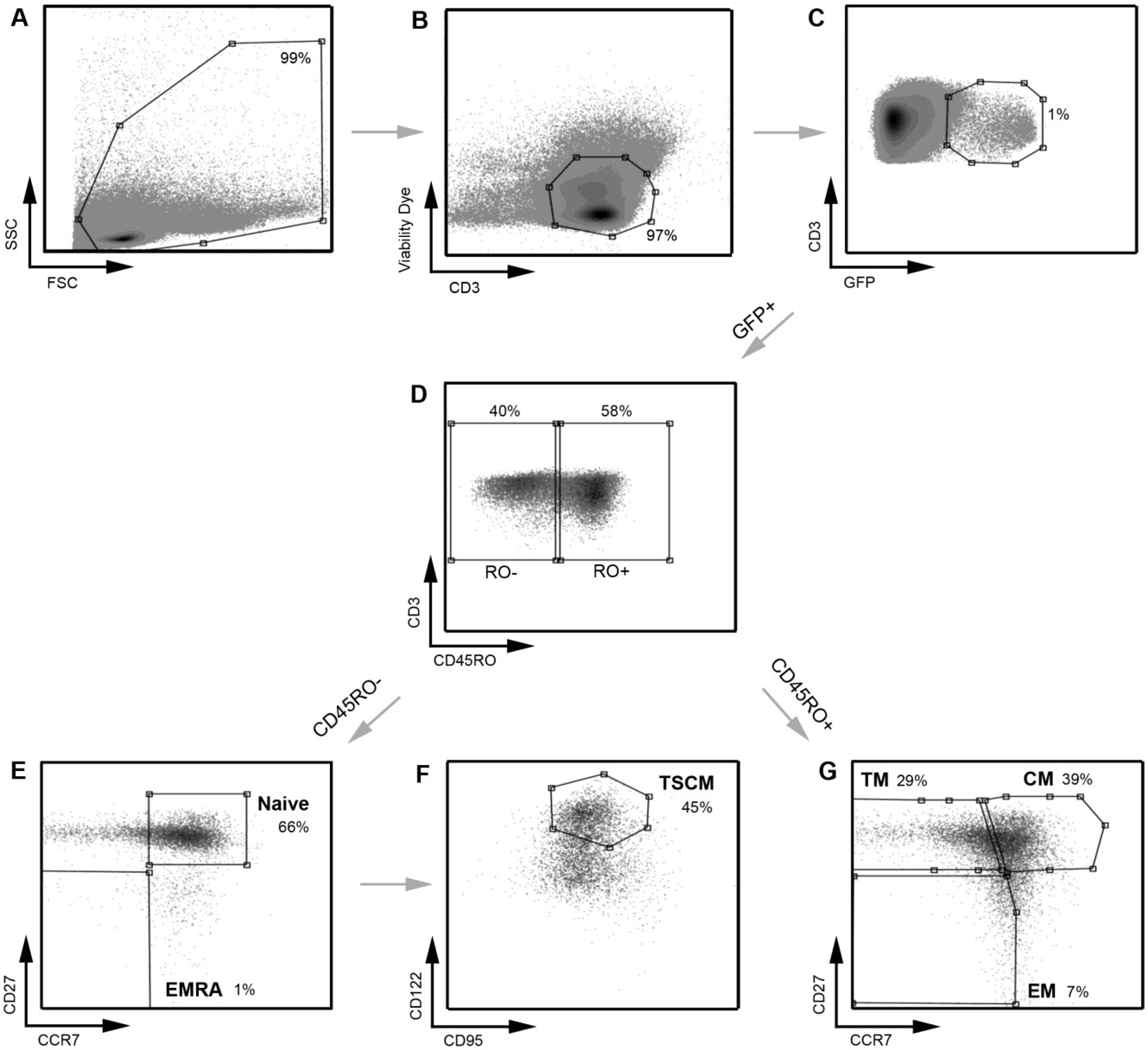
2.1. Development and Validation of a T Cell Assay for the Detection of Infected TSCM

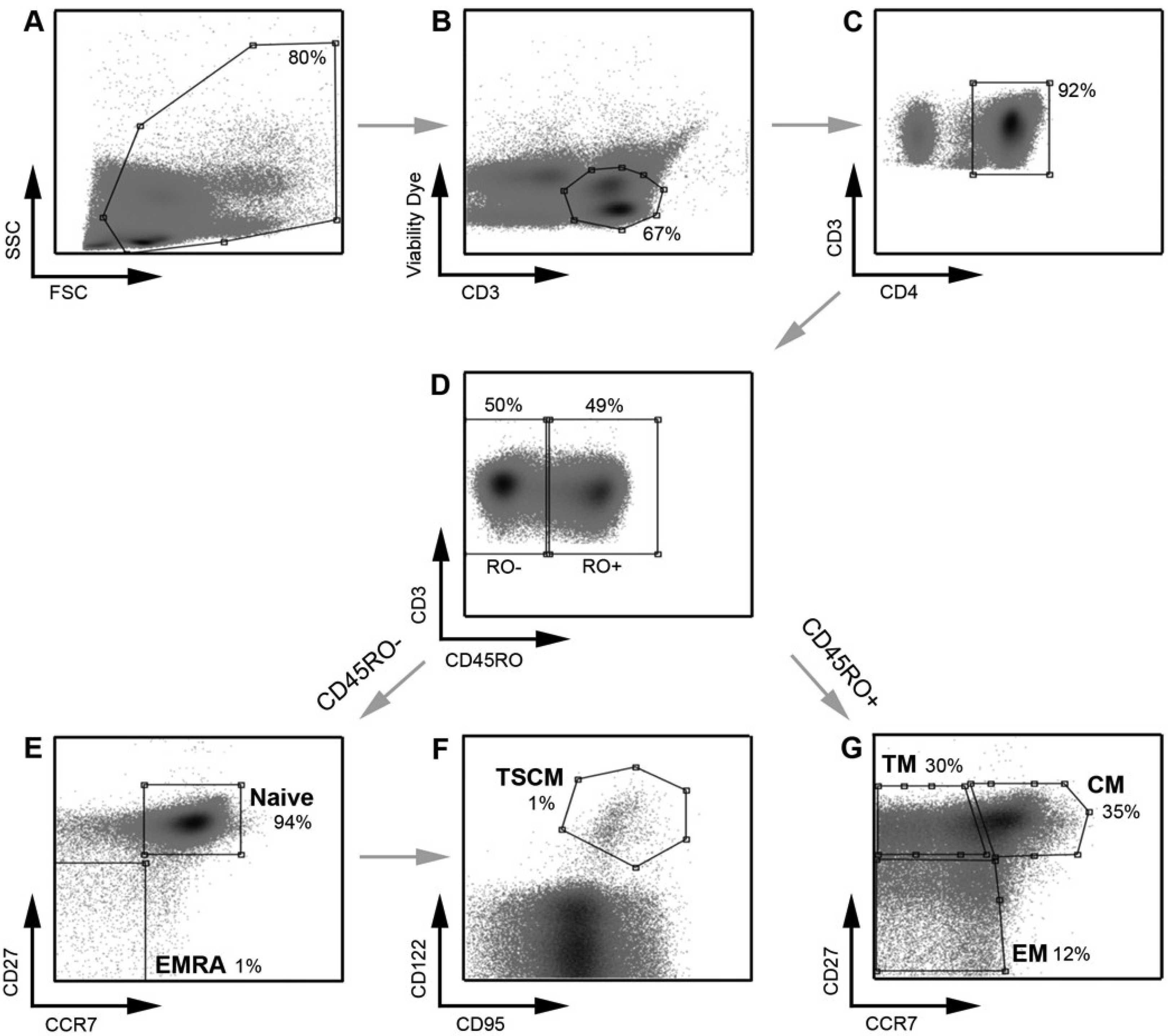{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Marker | Fluorochrome |
|---|---|
| CD4 | FITC |
| CD122 | EF710 (PerCPCy5.5) |
| CCR7 | AF-647 |
| CD3 | APCCy7 |
| CD45RO | EF450 (Pacific Blue) |
| Viability Dye * | EF506 (Amcyan) |
| CD95 | PE-CF594 |
| CD27 ^ | PE-Cy7 |
| CCR5 # | PE |
| CXCR4 # | PECy5 |



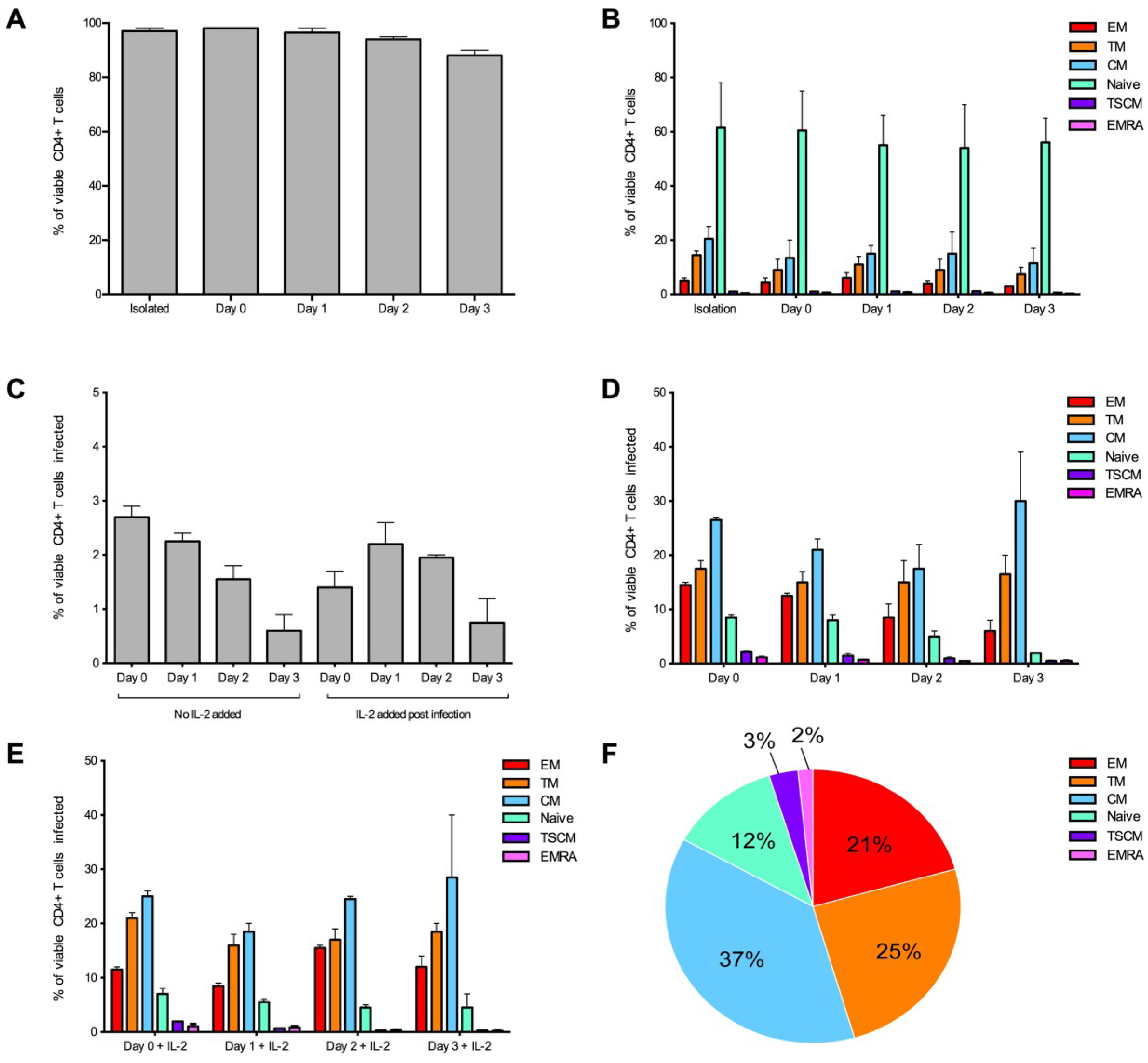
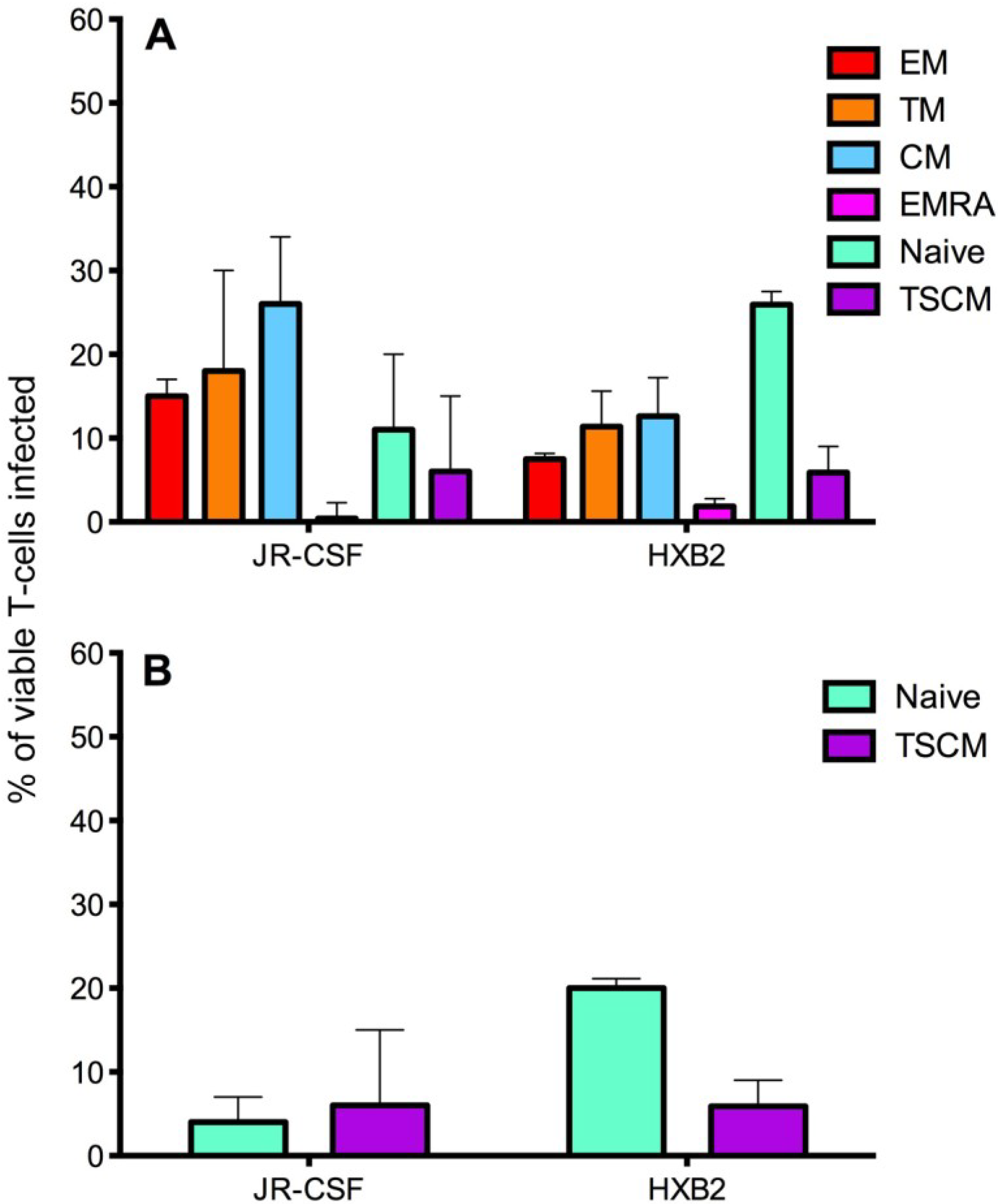
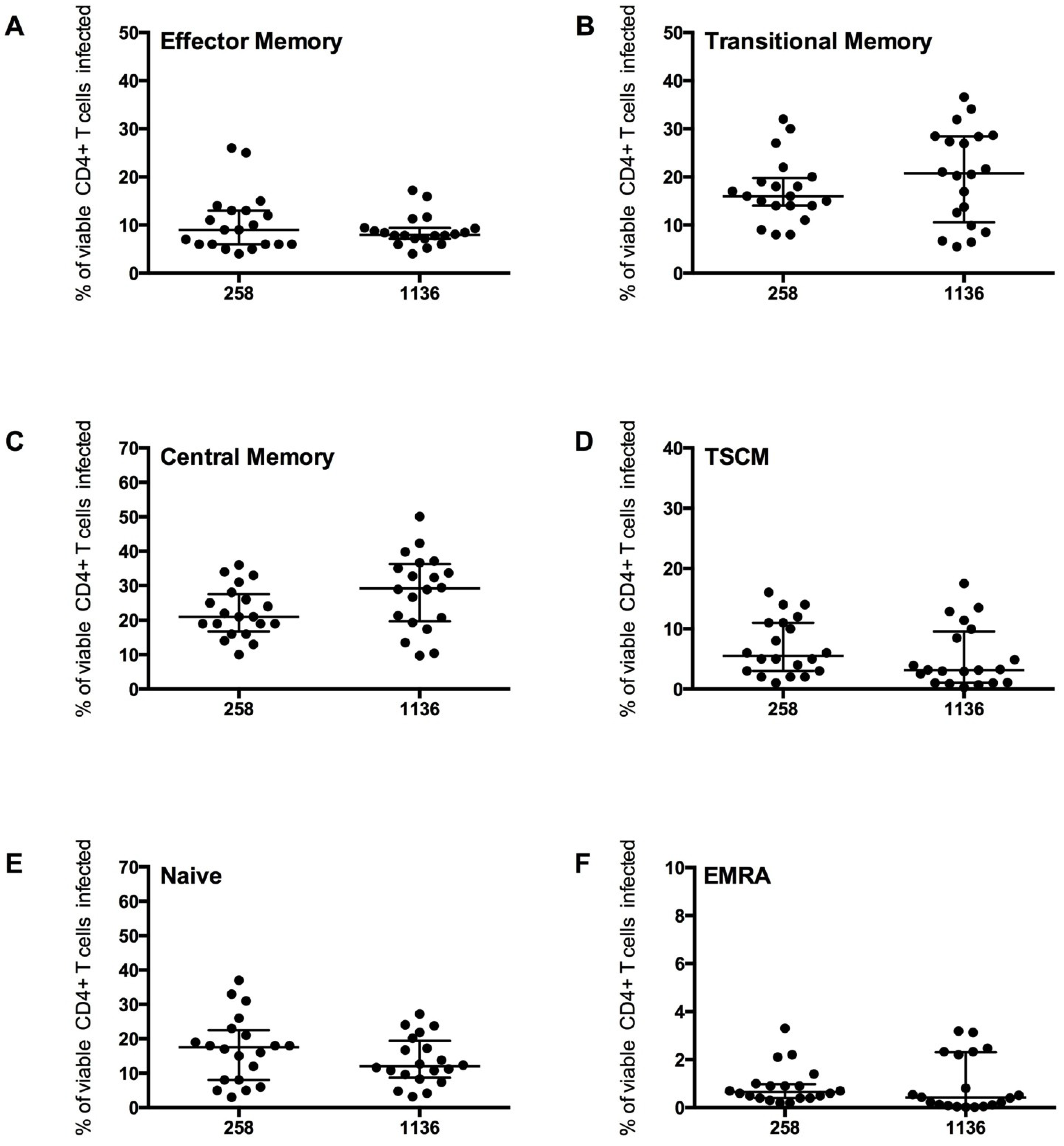
2.2. HIV-1 Infection in CD4+ T Cell Subsets by CCR5- and CXCR4-Using Viruses


2.3. Measurement of Infection in CD4+ T Cell Subsets by HIV-1 Subtype C Viruses

3. Experimental Section
3.1. Cells
3.2. HIV-1 Env Clones
3.3. Production and Quantitation of Env Pseudotyped GFP Reporter Viruses
3.4. Enumeration of HIV-1 Infection in CD4+ T Cell Subsets
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Gattinoni, L.; Restifo, N.P. Moving T memory stem cells to the clinic. Blood 2013, 121, 567–578. [Google Scholar] [CrossRef]
- Kalia, V.; Sarkar, S.; Ahmed, R. CD8 T-cell memory differentiation during acute and chronic viral infections. Adv. Exp. Med. Biol. 2010, 684, 79–95. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Ahmed, R. T-cell memory differentiation: Insights from transcriptional signatures and epigenetics. Immunology 2013, 139, 277–284. [Google Scholar] [CrossRef]
- Luckey, C.J.; Weaver, C.T. Stem-cell-like qualities of immune memory; CD4+ T cells join the party. Cell Stem Cell 2012, 10, 107–108. [Google Scholar] [CrossRef]
- Fritsch, R.D.; Shen, X.; Sims, G.P.; Hathcock, K.S.; Hodes, R.J.; Lipsky, P.E. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J. Immunol. 2005, 175, 6489–6497. [Google Scholar]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Ann. Rev. Immunol. 2004, 22, 745–763. [Google Scholar]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Geginat, J.; Sallusto, F.; Lanzavecchia, A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J. Exp. Med. 2001, 194, 1711–1719. [Google Scholar] [CrossRef]
- Riou, C.; Yassine-Diab, B.; Van grevenynghe, J.; Somogyi, R.; Greller, L.D.; Gagnon, D.; Gimmig, S.; Wilkinson, P.; Shi, Y.; Cameron, M.J.; et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J. Exp. Med. 2007, 204, 79–91. [Google Scholar] [CrossRef]
- Lugli, E.; Gattinoni, L.; Roberto, A.; Mavilio, D.; Price, D.A.; Restifo, N.P.; Roederer, M. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat. Protocol. 2013, 8, 33–42. [Google Scholar]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Sant, A.J.; McMichael, A. Revealing the role of CD4+ T cells in viral immunity. J. Exp. Med. 2012, 209, 1391–1395. [Google Scholar] [CrossRef]
- Hazenberg, M.D.; Otto, S.A.; van Benthem, B.H.; Roos, M.T.; Coutinho, R.A.; Lange, J.M.; Hamann, D.; Prins, M.; Miedema, F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS 2003, 17, 1881–1888. [Google Scholar] [CrossRef]
- Douek, D.C.; Picker, L.J.; Koup, R.A. T Cell Dynamics in HIV-1 Infection. Ann. Rev. Immunol. 2003, 21, 265–304. [Google Scholar] [CrossRef]
- Lugli, E.; Dominguez, M.; Gattinoni, L.; Chattopadhyay, P.; Bolton, D.; Song, K.; Klatt, N.; Brenchley, J.; Vaccari, M.; Gostick, E.; et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 2013, 123, 594–599. [Google Scholar]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Buzon, M. T memory stem cells: A long-term reservoir for HIV-1. In Proceedings of the ID Week 2012 Meeting, San Diego, CA, USA, 17–21 October 2012. Paper #594..
- Embretson, J.; Zupancic, M.; Ribas, J.; Burke, A.; Racz, P.; Tenner-Racz, K.; Haase, A. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 1993, 262, 359–362. [Google Scholar]
- Flynn, J.K.; Paukovics, G.; Moore, M.S.; Ellett, A.; Gray, L.R.; Duncan, R.; Salimi, H.; Jubb, B.; Westby, M.; Purcell, D.F.; et al. The magnitude of HIV-1 resistance to the CCR5 antagonist maraviroc may impart a differential alteration in HIV-1 tropism for macrophages and T-cell subsets. Virology 2013, 442, 51–58. [Google Scholar] [CrossRef]
- Perfetto, S.P.; Chattopadhyay, P.K.; Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 2004, 4, 648–655. [Google Scholar] [CrossRef]
- Appay, V.; van Lier, R.A.; Sallusto, F.; Roederer, M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A 2008, 73, 975–983. [Google Scholar]
- De Rosa, S.C.; Herzenberg, L.A.; Herzenberg, L.A.; Roederer, M. 11-color, 13-parameter flow cytometry: Identification of human naive T cells by phenotype, function, and T-cell receptor diversity. Nat. Med. 2001, 7, 245–248. [Google Scholar] [CrossRef]
- Roche, M.; Jakobsen, M.R.; Ellett, A.; Salimiseyedabad, H.; Jubb, B.; Westby, M.; Lee, B.; Lewin, S.R.; Churchill, M.J.; Gorry, P.R. HIV-1 predisposed to acquiring resistance to maraviroc (MVC) and other CCR5 antagonists in vitro has an inherent, low-level ability to utilize MVC-bound CCR5 for entry. Retrovirology 2011, 8, 89. [Google Scholar] [CrossRef]
- Roche, M.; Jakobsen, M.R.; Sterjovski, J.; Ellett, A.; Posta, F.; Lee, B.; Jubb, B.; Westby, M.; Lewin, S.R.; Ramsland, P.A.; et al. HIV-1 escape from the CCR5 antagonist maraviroc associated with an altered and less efficient mechanism of gp120-CCR5 engagement that attenuates macrophage-tropism. J. Virol. 2011, 85, 4330–4342. [Google Scholar] [CrossRef]
- Gorry, P.R.; Ancuta, P. Coreceptors and HIV-1 pathogenesis. Curr. HIV AIDS Rep. 2011, 8, 45–53. [Google Scholar] [CrossRef]
- Lee, B.; Sharron, M.; Montaner, L.J.; Weissman, D.; Doms, R.W. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 5215–5220. [Google Scholar] [CrossRef]
- Pfaff, J.M.; Wilen, C.B.; Harrison, J.E.; Demarest, J.F.; Lee, B.; Doms, R.W.; Tilton, J.C. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 2010, 84, 6505–6514. [Google Scholar] [CrossRef]
- Bjorndal, A.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyo, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487. [Google Scholar]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use coreceptor use correlates with disease progression in HIV-1—Infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef]
- Cashin, K.; Jakobsen, M.R.; Sterjovski, J.; Roche, M.; Ellett, A.; Flynn, J.K.; Borm, K.; Gouillou, M.; Churchill, M.J.; Gorry, P.R. Linkages between HIV-1 specificity for CCR5 or CXCR4 and in vitro usage of alternative coreceptors during progressive HIV-1 subtype C infection. Retrovirology 2013, 10, 98. [Google Scholar] [CrossRef]
- Jakobsen, M.; Cashin, K.; Roche, M.; Sterjovski, J.; Ellett, A.; Borm, K.; Flynn, J.; Erikstrup, C.; Gouillou, M.; Gray, L.; et al. Longitudinal analysis of CCR5 and CXCR4 usage in a cohort of antiretroviral therapy-naïve subjects with progressive HIV-1 subtype C infection. PLoS One 2013, 8, e65950. [Google Scholar] [CrossRef]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef]
- Gao, F.; Morrison, S.G.; Robertson, D.L.; Thornton, C.L.; Craig, S.; Karlsson, G.; Sodroski, J.; Morgado, M.; Galvao-Castro, B.; von Briesen, H.; et al. Molecular cloning and analysis of functional envelope genes from human immunodeficiency virus type 1 sequence subtypes A through G. The WHO and NIAID Networks for HIV Isolation and Characterization. J. Virol. 1996, 70, 1651–1667. [Google Scholar]
- Cashin, K.; Gray, L.R.; Jakobsen, M.R.; Sterjovski, J.; Churchill, M.J.; Gorry, P.R. CoRSeqV3-C: a novel HIV-1 subtype C specific V3 sequence based coreceptor usage prediction algorithm. Retrovirology 2013, 10, 24. [Google Scholar] [CrossRef]
- Center, R.J.; Wheatley, A.K.; Campbell, S.M.; Gaeguta, A.J.; Peut, V.; Alcantara, S.; Siebentritt, C.; Kent, S.J.; Purcell, D.F. Induction of HIV-1 subtype B and AE-specific neutralizing antibodies in mice and macaques with DNA prime and recombinant gp140 protein boost regimens. Vaccine 2009, 27, 6605–6612. [Google Scholar] [CrossRef]
- Yap, S.H.; Sheen, C.W.; Fahey, J.; Zanin, M.; Tyssen, D.; Lima, V.D.; Wynhoven, B.; Kuiper, M.; Sluis-Cremer, N.; Harrigan, P.R.; et al. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 2007, 4, e335. [Google Scholar] [CrossRef] [Green Version]
- BD Biosciences. Multicolor Flow Cytometry Absorption and Emission Spectra. Available online: http://www.bdbiosciences.com/research/multicolor/spectrumguide/ (accessed on 10 February 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flynn, J.K.; Paukovics, G.; Cashin, K.; Borm, K.; Ellett, A.; Roche, M.; Jakobsen, M.R.; Churchill, M.J.; Gorry, P.R. Quantifying Susceptibility of CD4+ Stem Memory T-Cells to Infection by Laboratory Adapted and Clinical HIV-1 Strains. Viruses 2014, 6, 709-726. https://doi.org/10.3390/v6020709
Flynn JK, Paukovics G, Cashin K, Borm K, Ellett A, Roche M, Jakobsen MR, Churchill MJ, Gorry PR. Quantifying Susceptibility of CD4+ Stem Memory T-Cells to Infection by Laboratory Adapted and Clinical HIV-1 Strains. Viruses. 2014; 6(2):709-726. https://doi.org/10.3390/v6020709
Chicago/Turabian StyleFlynn, Jacqueline K., Geza Paukovics, Kieran Cashin, Katharina Borm, Anne Ellett, Michael Roche, Martin R. Jakobsen, Melissa J. Churchill, and Paul R. Gorry. 2014. "Quantifying Susceptibility of CD4+ Stem Memory T-Cells to Infection by Laboratory Adapted and Clinical HIV-1 Strains" Viruses 6, no. 2: 709-726. https://doi.org/10.3390/v6020709
APA StyleFlynn, J. K., Paukovics, G., Cashin, K., Borm, K., Ellett, A., Roche, M., Jakobsen, M. R., Churchill, M. J., & Gorry, P. R. (2014). Quantifying Susceptibility of CD4+ Stem Memory T-Cells to Infection by Laboratory Adapted and Clinical HIV-1 Strains. Viruses, 6(2), 709-726. https://doi.org/10.3390/v6020709