The DNA Damage Response Induced by Infection with Human Cytomegalovirus and Other Viruses

Abstract
:1. Introduction
2. HCMV
3. Cell Cycle Checkpoints
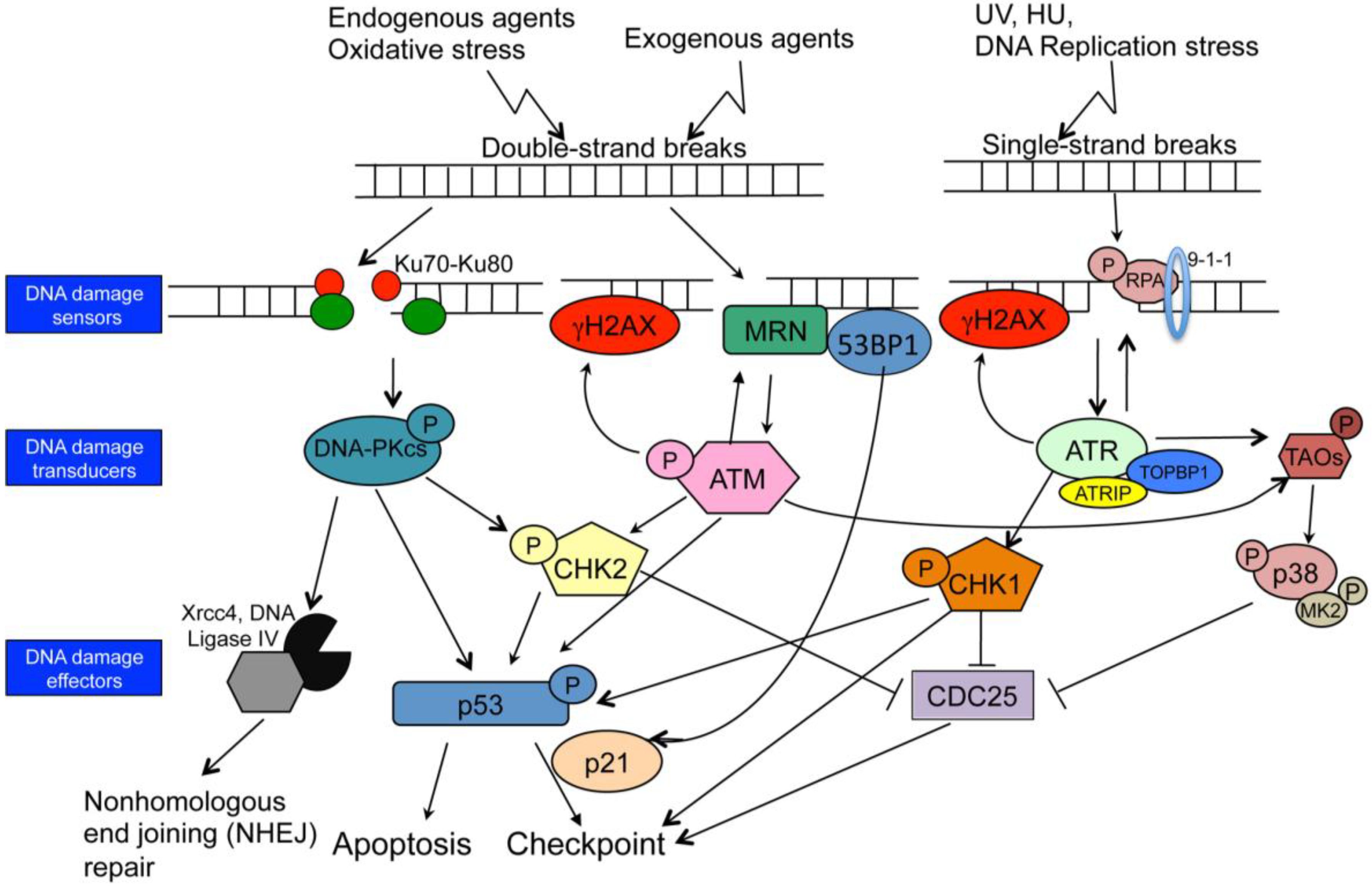
4. DNA Damage Response (DDR)

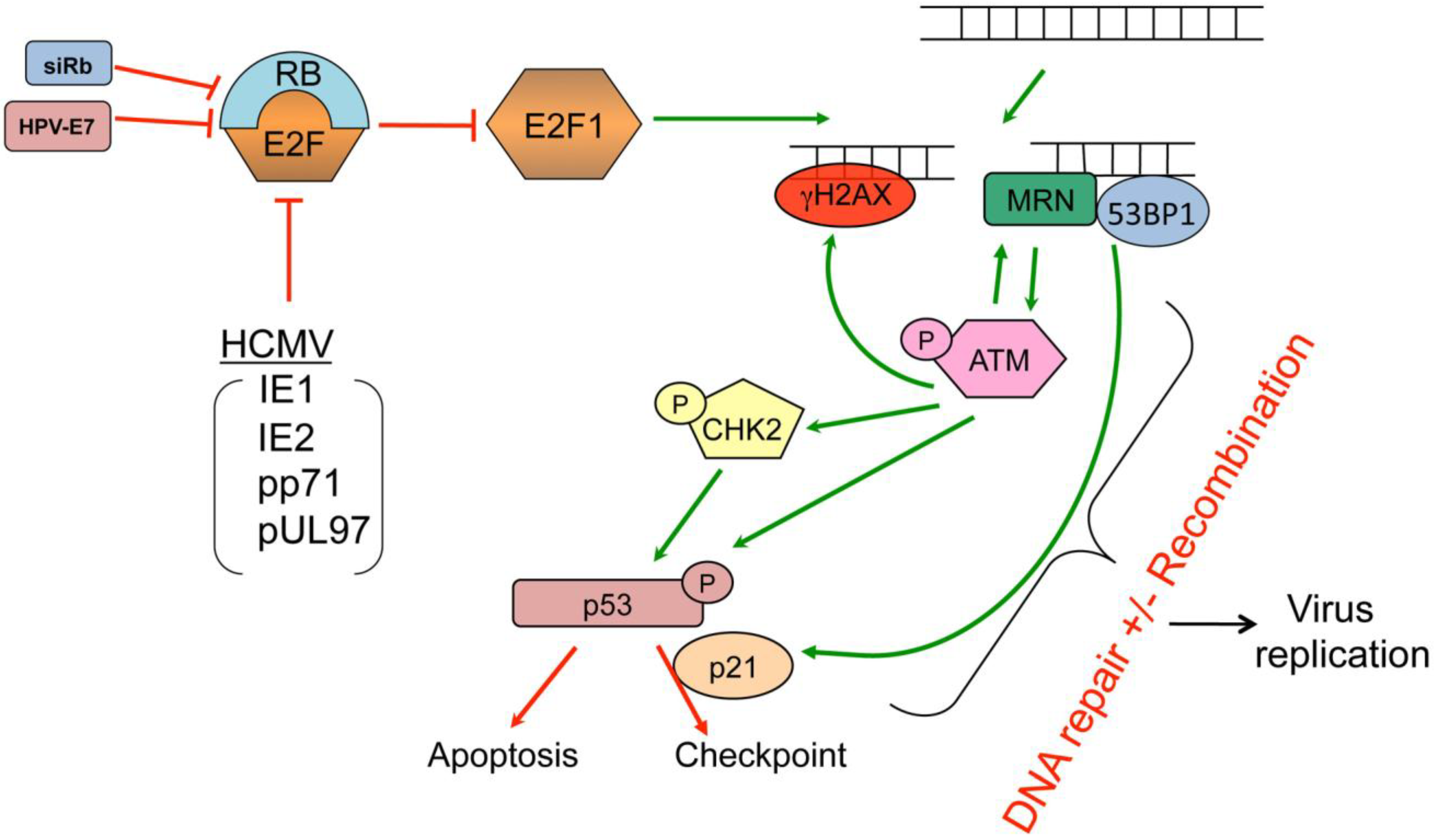
5. The pRB-E2F Complex
6. The Link between Cell Cycle and DDR
6.1. CHK1 and CHK2 Kinases Control the Cell Cycle in Response to DNA Damage
6.2. A Novel Cell Cycle Checkpoint Kinase Pathway, MK2, that also Induced Cell Cycle Arrest
7. Many Viruses can Induce DNA Damage Responses and Modulate Cell Cycle Progression

{kind=link}
{kind=link}
{kind=link}
| Virus that Induce DNA damage response (DDR) | Abbreviation | Virus type | DDR factors activated | DDR factors required for virus replication | References |
|---|---|---|---|---|---|
| Human cytomegalovirus | HCMV | dsDNA, β-herpesvirus | ATM, CHK2, p53, H2AX NBS1, CHK1 | ATM, p53, H2AX | [94,98] |
| Herpes simplex virus type 1 | HSV-1 | dsDNA, α-herpesvirus | ATM, CHK2, 53BP1, NBS1 | ATM, Mre11 | [95] |
| Epstein-Barr virus | EBV | dsDNA, γ-herpesvirus | ATM, CHK2, Nbs1, H2AX, p53, CHK1 | XPC | [96,99] |
| Murine gammaherpesvirus 68 | γHV68 | dsDNA, γ-herpesvirus | ATM, H2AX, p53, CHK1 | ATM, H2AX | [100,101] |
| Simian virus type 40 | SV40 | dsDNA, polyomavirus | ATM, CHK1, CHK2, p53 | ATM, Rad51, FancD2 | [97,102,103] |
| Human papillomavirus | HPV | dsDNA, papillomavirus | ATM, CHK2, H2AX, NBS1, CHK1, BRCA1 | ATM, CHK2 | [104,105,106,107,108] |
| Human parvovirus B19 | B19V | ssDNA, parvovirus | ATM, CHK2, ATR, DNA-PKcs, CHK1, Ku70/Ku80, H2AX, RPA-32 | ATR, CHK1, DNA-PKcs, Ku70/ku80 | [109,110,111] |
| Adeno-associated virus | AAV | ssDNA, parvovirus | ATM, CHK2, DNA-PKcs, SMC1, H2AX, CHK1, RPA32 | DNA-Pkcs | [112,113] |
| Human T-cell lymphotrophic virus type 1 | HTLV1 | ssRNA/dsDNA, retrovirus | ATM, CHK2, H2AX, NBS1, DNA-PKcs | N/A | [114,115,116,117,118,119] |
| Human immunodeficiency virus type 1 | HIV-1 | ssRNA/dsDNA, lentivirus | ATM, H2AX, p53, NBS1, ATR, CHK1, P38MAPK | ATM | [120,121,122] |
| Rift Valley fever virus | RVFV | ssRNA, arbovirus | ATM, CHK2, H2AX, p53 | ATM, CHK2, p53 | [123,124] |
| Hepatitis C virus | HCV | ssRNA, flavivirus | ATM, CHK2, H2AX, CHK1 | ATM, CHK2 | [125,126] |
8. HCMV Modulates the Cell Cycle and Checkpoints


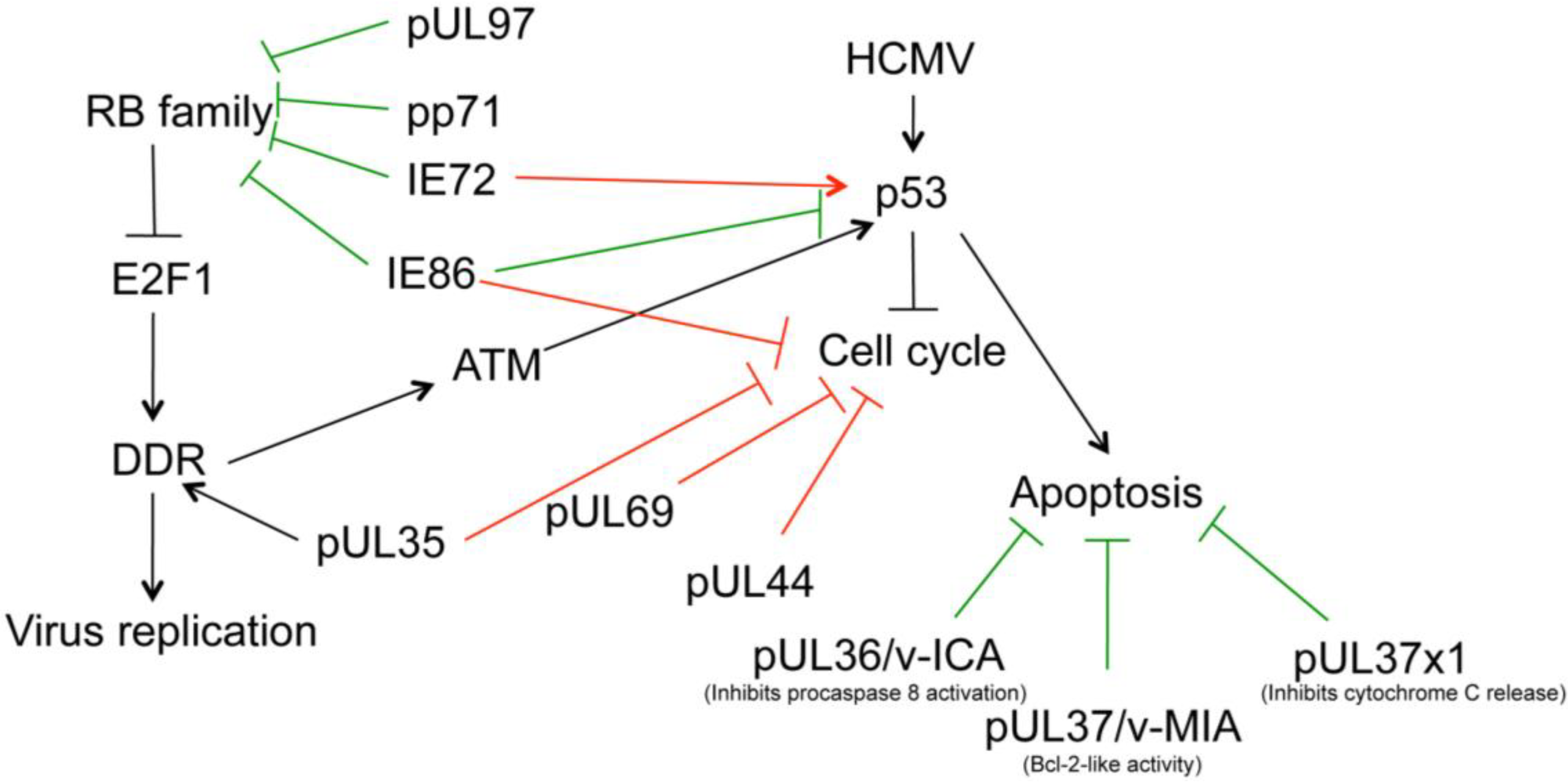
9. HCMV, Deregulation of the Cell Cycle, and DNA Damage Signaling
9.1. IE1 Can Inactivate p107 and p130
9.2. IE2 Binds pRB and p53
9.3. pp71 Binds to pRB, p107 and p130
9.4. pUL97 Phosphorylates RB Protein
9.5. pUL69 Modulates CDK Function
9.6. E2F1-mediated DNA Damage Response
10. Which DDR Factors Contribute to HCMV Replication?
11. Difference and Similarities in the DDR Induced by HCMV and Other Viruses
11.1. HCMV Is Similar to Other Viruses that Use DDR (ATM Signaling) for Replication
11.2. HCMV is Different from Other Viruses Not Using ATM or that Block DDR for Replication
12. HCMV Infection Results in the Relocalization of DDR Proteins to Virus Replication Compartments (RCs)
13. Does DNA Damage Exist in Cellular or Viral DNA During Infection?
14. Future Perspectives
Acknowledgments
Authors Contributions
Conflicts of Interest
References and Notes
- Vancikova, Z.; Dvorak, P. Cytomegalovirus infection in immunocompetent and immunocompromised individuals—A review. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2001, 1, 179–187. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126, Table of Contents. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Walter, S. Cytomegalovirus. Curr. Opin. Infect. Dis. 2005, 18, 241–245. [Google Scholar] [CrossRef]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; McGregor Dallas, S.R.; Smit, M.; Soroceanu, L.; Cobbs, C.S. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012, 14, 246–255. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd; Kouzarides, T.; Martignetti, J.A.; et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar]
- Rigoutsos, I.; Novotny, J.; Huynh, T.; Chin-Bow, S.T.; Parida, L.; Platt, D.; Coleman, D.; Shenk, T. In silico pattern-based analysis of the human cytomegalovirus genome. J. Virol. 2003, 77, 4326–4344. [Google Scholar]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 2011, 7, e1001344. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef]
- Baldick, C.J., Jr.; Shenk, T. Proteins associated with purified human cytomegalovirus particles. J. Virol. 1996, 70, 6097–6105. [Google Scholar]
- Hayashi, M.L.; Blankenship, C.; Shenk, T. Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 2692–2696. [Google Scholar] [CrossRef]
- DeMarchi, J.M.; Schmidt, C.A.; Kaplan, A.S. Patterns of transcription of human cytomegalovirus in permissively infected cells. J. Virol. 1980, 35, 277–286. [Google Scholar]
- Wathen, M.W.; Stinski, M.F. Temporal patterns of human cytomegalovirus transcription: Mapping the viral RNAs synthesized at immediate early, early, and late times after infection. J. Virol. 1982, 41, 462–477. [Google Scholar]
- Stamminger, T.; Fleckenstein, B. Immediate-early transcription regulation of human cytomegalovirus. Curr. Top. Microbiol. Immunol. 1990, 154, 3–19. [Google Scholar]
- Fortunato, E.A.; Sommer, M.H.; Yoder, K.; Spector, D.H. Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J. Virol. 1997, 71, 8176–8185. [Google Scholar]
- Poma, E.E.; Kowalik, T.F.; Zhu, L.; Sinclair, J.H.; Huang, E.S. The human cytomegalovirus IE1–72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol. 1996, 70, 7867–7877. [Google Scholar]
- Fortunato, E.A.; Spector, D.H. Regulation of human cytomegalovirus gene expression. Adv. Virus Res. 1999, 54, 61–128. [Google Scholar] [CrossRef]
- Penfold, M.E.; Mocarski, E.S. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 1997, 239, 46–61. [Google Scholar]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Soderberg-Naucler, C.; Streblow, D.N.; Fish, K.N.; Allan-Yorke, J.; Smith, P.P.; Nelson, J.A. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol. 2001, 75, 7543–7554. [Google Scholar]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J. Clin. Invest. 1997, 100, 3154–3163. [Google Scholar] [CrossRef]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef]
- Petrucelli, A.; Rak, M.; Grainger, L.; Goodrum, F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 2009, 83, 5615–5629. [Google Scholar] [CrossRef]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef]
- Mason, G.M.; Poole, E.; Sissons, J.G.; Wills, M.R.; Sinclair, J.H. Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc. Natl. Acad. Sci. USA 2012, 109, 14538–14543. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef]
- Elledge, S.J. Cell cycle checkpoints: Preventing an identity crisis. Science 1996, 274, 1664–1672. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Gennery, A.R.; Seidel, J.; Concannon, P.; Jeggo, P.A. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst) 2004, 3, 1227–1235. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Taylor, A.M.; Groom, A.; Byrd, P.J. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst) 2004, 3, 1219–1225. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Shiloh, Y.; Kastan, M.B. ATM: Genome stability, neuronal development, and cancer cross paths. Adv. Cancer Res. 2001, 83, 209–254. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Rasheed, N.; Hu, Y.; Boast, S.; Ishii, T.; Nakayama, K.; Nakayama, K.I.; Goff, S.P. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev. 2004, 18, 1824–1837. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Nevins, J.R. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar]
- Dyson, N. pRB, p107 and the regulation of the E2F transcription factor. J. Cell. Sci. Suppl. 1994, 18, 81–87. [Google Scholar] [CrossRef]
- La Thangue, N.B. DP and E2F proteins: Components of a heterodimeric transcription factor implicated in cell cycle control. Curr. Opin. Cell Biol. 1994, 6, 443–450. [Google Scholar] [CrossRef]
- La Thangue, N.B. E2F and the molecular mechanisms of early cell-cycle control. Biochem. Soc. Trans. 1996, 24, 54–59. [Google Scholar]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar] [CrossRef]
- Helin, K.; Lees, J.A.; Vidal, M.; Dyson, N.; Harlow, E.; Fattaey, A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 1992, 70, 337–350. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Krek, W.; Sellers, W.R.; DeCaprio, J.A.; Ajchenbaum, F.; Fuchs, C.S.; Chittenden, T.; Li, Y.; Farnham, P.J.; Blanar, M.A.; et al. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell 1992, 70, 351–364. [Google Scholar] [CrossRef]
- Qin, X.Q.; Livingston, D.M.; Ewen, M.; Sellers, W.R.; Arany, Z.; Kaelin, W.G., Jr. The transcription factor E2F-1 is a downstream target of RB action. Mol. Cell. Biol. 1995, 15, 742–755. [Google Scholar]
- Ikeda, M.A.; Jakoi, L.; Nevins, J.R. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 3215–3220. [Google Scholar] [CrossRef]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar]
- Nevins, J.R. Disruption of cell-cycle control by viral oncoproteins. Biochem. Soc. Trans. 1993, 21, 935–938. [Google Scholar]
- Nevins, J.R. Cell cycle targets of the DNA tumor viruses. Curr. Opin. Genet. Dev. 1994, 4, 130–134. [Google Scholar]
- Moberg, K.; Starz, M.A.; Lees, J.A. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell. Biol. 1996, 16, 1436–1449. [Google Scholar]
- Ginsberg, D.; Vairo, G.; Chittenden, T.; Xiao, Z.X.; Xu, G.; Wydner, K.L.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994, 8, 2665–2679. [Google Scholar] [CrossRef]
- Hijmans, E.M.; Voorhoeve, P.M.; Beijersbergen, R.L.; van’t Veer, L.J.; Bernards, R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol. Cell. Biol. 1995, 15, 3082–3089. [Google Scholar]
- Vairo, G.; Livingston, D.M.; Ginsberg, D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995, 9, 869–881. [Google Scholar] [CrossRef]
- Beijersbergen, R.L.; Kerkhoven, R.M.; Zhu, L.; Carlee, L.; Voorhoeve, P.M.; Bernards, R. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes Dev. 1994, 8, 2680–2690. [Google Scholar] [CrossRef]
- Morkel, M.; Wenkel, J.; Bannister, A.J.; Kouzarides, T.; Hagemeier, C. An E2F-like repressor of transcription. Nature 1997, 390, 567–568. [Google Scholar] [CrossRef]
- Cartwright, P.; Muller, H.; Wagener, C.; Holm, K.; Helin, K. E2F-6: A novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 1998, 17, 611–623. [Google Scholar]
- Gaubatz, S.; Wood, J.G.; Livingston, D.M. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc. Natl. Acad. Sci. USA 1998, 95, 9190–9195. [Google Scholar] [CrossRef]
- Trimarchi, J.M.; Fairchild, B.; Verona, R.; Moberg, K.; Andon, N.; Lees, J.A. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl. Acad. Sci. USA 1998, 95, 2850–2855. [Google Scholar]
- de Bruin, A.; Maiti, B.; Jakoi, L.; Timmers, C.; Buerki, R.; Leone, G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 2003, 278, 42041–42049. [Google Scholar]
- Di Stefano, L.; Jensen, M.R.; Helin, K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003, 22, 6289–6298. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef]
- Lukas, C.; Bartkova, J.; Latella, L.; Falck, J.; Mailand, N.; Schroeder, T.; Sehested, M.; Lukas, J.; Bartek, J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001, 61, 4990–4993. [Google Scholar]
- Hlavova, M.; Cizkova, M.; Vitova, M.; Bisova, K.; Zachleder, V. DNA damage during G2 phase does not affect cell cycle progression of the green alga Scenedesmus quadricauda. PLoS One 2011, 6, e19626. [Google Scholar]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar]
- Raman, M.; Earnest, S.; Zhang, K.; Zhao, Y.; Cobb, M.H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007, 26, 2005–2014. [Google Scholar] [CrossRef]
- Hirose, Y.; Katayama, M.; Stokoe, D.; Haas-Kogan, D.A.; Berger, M.S.; Pieper, R.O. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol. Cell. Biol. 2003, 23, 8306–8315. [Google Scholar] [CrossRef]
- Mikhailov, A.; Shinohara, M.; Rieder, C.L. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 2004, 166, 517–526. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Lazzaro, F.; Giannattasio, M.; Puddu, F.; Granata, M.; Pellicioli, A.; Plevani, P.; Muzi-Falconi, M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst) 2009, 8, 1055–1067. [Google Scholar] [CrossRef]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or abusing: Viruses and the cellular DNA damage response. Trends Microbiol. 2007, 15, 119–126. [Google Scholar] [CrossRef]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar]
- Nikitin, P.A.; Luftig, M.A. At a crossroads: Human DNA tumor viruses and the host DNA damage response. Future Virol. 2011, 6, 813–830. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Chaurushiya, M.S.; Weitzman, M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst) 2009, 8, 1166–1176. [Google Scholar] [CrossRef]
- Flemington, E.K. Herpesvirus lytic replication and the cell cycle: Arresting new developments. J. Virol. 2001, 75, 4475–4481. [Google Scholar] [CrossRef]
- Nascimento, R.; Costa, H.; Parkhouse, R.M. Virus manipulation of cell cycle. Protoplasma 2012, 249, 519–528. [Google Scholar] [CrossRef]
- E, X.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef]
- Lu, C.C.; Chen, Y.C.; Wang, J.T.; Yang, P.W.; Chen, M.R. Xeroderma pigmentosum C is involved in Epstein Barr virus DNA replication. J. Gen. Virol. 2007, 88, 3234–3243. [Google Scholar] [CrossRef]
- Boichuk, S.; Hu, L.; Hein, J.; Gjoerup, O.V. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J. Virol. 2010, 84, 8007–8020. [Google Scholar]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef]
- Kudoh, A.; Fujita, M.; Zhang, L.; Shirata, N.; Daikoku, T.; Sugaya, Y.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 2005, 280, 8156–8163. [Google Scholar] [CrossRef]
- Liang, X.; Pickering, M.T.; Cho, N.H.; Chang, H.; Volkert, M.R.; Kowalik, T.F.; Jung, J.U. Deregulation of DNA damage signal transduction by herpesvirus latency-associated M2. J. Virol. 2006, 80, 5862–5874. [Google Scholar] [CrossRef]
- Tarakanova, V.L.; Leung-Pineda, V.; Hwang, S.; Yang, C.W.; Matatall, K.; Basson, M.; Sun, R.; Piwnica-Worms, H.; Sleckman, B.P.; Virgin, H.W. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 2007, 1, 275–286. [Google Scholar] [CrossRef]
- Shi, Y.; Dodson, G.E.; Shaikh, S.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar] [CrossRef]
- Zhao, X.; Madden-Fuentes, R.J.; Lou, B.X.; Pipas, J.M.; Gerhardt, J.; Rigell, C.J.; Fanning, E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 2008, 82, 5316–5328. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Lou, S.; Luo, Y.; Cheng, F.; Huang, Q.; Shen, W.; Kleiboeker, S.; Tisdale, J.F.; Liu, Z.; Qiu, J. Human parvovirus B19 DNA replication induces a DNA damage response that is dispensable for cell cycle arrest at phase G2/M. J. Virol. 2012, 86, 10748–10758. [Google Scholar] [CrossRef]
- Luo, Y.; Kleiboeker, S.; Deng, X.; Qiu, J. Human parvovirus B19 infection causes cell cycle arrest of human erythroid progenitors at late S phase that favors viral DNA replication. J. Virol. 2013, 87, 12766–12775. [Google Scholar]
- Luo, Y.; Lou, S.; Deng, X.; Liu, Z.; Li, Y.; Kleiboeker, S.; Qiu, J. Parvovirus B19 infection of human primary erythroid progenitor cells triggers ATR-Chk1 signaling, which promotes B19 virus replication. J. Virol. 2011, 85, 8046–8055. [Google Scholar] [CrossRef]
- Choi, Y.K.; Nash, K.; Byrne, B.J.; Muzyczka, N.; Song, S. The effect of DNA-dependent protein kinase on adeno-associated virus replication. PLoS One 2010, 5, e15073. [Google Scholar]
- Schwartz, R.A.; Carson, C.T.; Schuberth, C.; Weitzman, M.D. Adeno-associated virus replication induces a DNA damage response coordinated by DNA-dependent protein kinase. J. Virol. 2009, 83, 6269–6278. [Google Scholar] [CrossRef]
- Baydoun, H.H.; Bai, X.T.; Shelton, S.; Nicot, C. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS One 2012, 7, e42226. [Google Scholar]
- Belgnaoui, S.M.; Fryrear, K.A.; Nyalwidhe, J.O.; Guo, X.; Semmes, O.J. The viral oncoprotein tax sequesters DNA damage response factors by tethering MDC1 to chromatin. J. Biol. Chem. 2010, 285, 32897–32905. [Google Scholar]
- Chandhasin, C.; Ducu, R.I.; Berkovich, E.; Kastan, M.B.; Marriott, S.J. Human T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage response. J. Virol. 2008, 82, 6952–6961. [Google Scholar] [CrossRef]
- Chlichlia, K.; Khazaie, K. HTLV-1 Tax: Linking transformation, DNA damage and apoptotic T-cell death. Chem. Biol. Interact. 2010, 188, 359–365. [Google Scholar] [CrossRef]
- Durkin, S.S.; Guo, X.; Fryrear, K.A.; Mihaylova, V.T.; Gupta, S.K.; Belgnaoui, S.M.; Haoudi, A.; Kupfer, G.M.; Semmes, O.J. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 2008, 283, 36311–36320. [Google Scholar] [CrossRef]
- Kinjo, T.; Ham-Terhune, J.; Peloponese, J.M., Jr.; Jeang, K.T. Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J. Virol. 2010, 84, 5431–5437. [Google Scholar]
- Ariumi, Y.; Trono, D. Ataxia-telangiectasia-mutated (ATM) protein can enhance human immunodeficiency virus type 1 replication by stimulating Rev function. J. Virol. 2006, 80, 2445–2452. [Google Scholar] [CrossRef]
- Lau, A.; Swinbank, K.M.; Ahmed, P.S.; Taylor, D.L.; Jackson, S.P.; Smith, G.C.; O’Connor, M.J. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 2005, 7, 493–500. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Nardacci, R.; Bourouba, M.; Subra, F.; Gros, L.; Seror, C.; Manic, G.; Rosselli, F.; Amendola, A.; Masdehors, P.; et al. Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia. PLoS One 2008, 3, e2458. [Google Scholar] [CrossRef]
- Austin, D.; Baer, A.; Lundberg, L.; Shafagati, N.; Schoonmaker, A.; Narayanan, A.; Popova, T.; Panthier, J.J.; Kashanchi, F.; Bailey, C.; et al. p53 Activation following Rift Valley fever virus infection contributes to cell death and viral production. PLoS One 2012, 7, e36327. [Google Scholar] [CrossRef]
- Baer, A.; Austin, D.; Narayanan, A.; Popova, T.; Kainulainen, M.; Bailey, C.; Kashanchi, F.; Weber, F.; Kehn-Hall, K. Induction of DNA Damage Signaling upon Rift Valley Fever Virus Infection Results in Cell Cycle Arrest and Increased Viral Replication. J. Biol. Chem. 2012, 287, 7399–7410. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Dansako, H.; Abe, K.; Ikeda, M.; Wakita, T.; Kato, N. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J. Virol. 2008, 82, 9639–9646. [Google Scholar] [CrossRef]
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis C virus infection downregulates the ligands of the activating receptor NKG2D. Cell. Mol. Immunol. 2008, 5, 475–478. [Google Scholar]
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 1999, 73, 650–657. [Google Scholar]
- Wilkinson, D.E.; Weller, S.K. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef]
- Bittar, C.; Shrivastava, S.; Bhanja Chowdhury, J.; Rahal, P.; Ray, R.B. Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS One 2013, 8, e62581. [Google Scholar]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 2010, 185, 6985–6998. [Google Scholar] [CrossRef]
- Forrester, N.A.; Sedgwick, G.G.; Thomas, A.; Blackford, A.N.; Speiseder, T.; Dobner, T.; Byrd, P.J.; Stewart, G.S.; Turnell, A.S.; Grand, R.J. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J. Virol. 2011, 85, 2201–2211. [Google Scholar] [CrossRef]
- Karen, K.A.; Hoey, P.J.; Young, C.S.; Hearing, P. Temporal regulation of the Mre11-Rad50-Nbs1 complex during adenovirus infection. J. Virol. 2009, 83, 4565–4573. [Google Scholar] [CrossRef]
- Evans, J.D.; Hearing, P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 2005, 79, 6207–6215. [Google Scholar] [CrossRef]
- Carson, C.T.; Orazio, N.I.; Lee, D.V.; Suh, J.; Bekker-Jensen, S.; Araujo, F.D.; Lakdawala, S.S.; Lilley, C.E.; Bartek, J.; Lukas, J.; et al. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009, 28, 652–662. [Google Scholar] [CrossRef]
- Liu, Y.; Shevchenko, A.; Berk, A.J. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 2005, 79, 14004–14016. [Google Scholar] [CrossRef]
- Araujo, F.D.; Stracker, T.H.; Carson, C.T.; Lee, D.V.; Weitzman, M.D. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 2005, 79, 11382–11391. [Google Scholar] [CrossRef]
- Mathew, S.S.; Bridge, E. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 2007, 365, 346–355. [Google Scholar] [CrossRef]
- Lakdawala, S.S.; Schwartz, R.A.; Ferenchak, K.; Carson, C.T.; McSharry, B.P.; Wilkinson, G.W.; Weitzman, M.D. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 2008, 82, 8362–8372. [Google Scholar] [CrossRef]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Spector, D.H. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 1998, 72, 2033–2039. [Google Scholar]
- Fortunato, E.A.; McElroy, A.K.; Sanchez, I.; Spector, D.H. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 2000, 8, 111–119. [Google Scholar] [CrossRef]
- Kalejta, R.F.; Shenk, T. Manipulation of the cell cycle by human cytomegalovirus. Front. Biosci. 2002, 7, d295–d306. [Google Scholar]
- Biswas, N.; Sanchez, V.; Spector, D.H. Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication. J. Virol. 2003, 77, 2369–2376. [Google Scholar] [CrossRef]
- Wiebusch, L.; Uecker, R.; Hagemeier, C. Human cytomegalovirus prevents replication licensing by inhibiting MCM loading onto chromatin. EMBO Rep. 2003, 4, 42–46. [Google Scholar] [CrossRef]
- Bresnahan, W.A.; Boldogh, I.; Thompson, E.A.; Albrecht, T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 1996, 224, 150–160. [Google Scholar] [CrossRef]
- Dittmer, D.; Mocarski, E.S. Human cytomegalovirus infection inhibits G1/S transition. J. Virol. 1997, 71, 1629–1634. [Google Scholar]
- Lu, M.; Shenk, T. Human cytomegalovirus UL69 protein induces cells to accumulate in G1 phase of the cell cycle. J. Virol. 1999, 73, 676–683. [Google Scholar]
- Kalejta, R.F.; Bechtel, J.T.; Shenk, T. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 2003, 23, 1885–1895. [Google Scholar] [CrossRef]
- Hume, A.J.; Finkel, J.S.; Kamil, J.P.; Coen, D.M.; Culbertson, M.R.; Kalejta, R.F. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 2008, 320, 797–799. [Google Scholar] [CrossRef]
- Salsman, J.; Jagannathan, M.; Paladino, P.; Chan, P.K.; Dellaire, G.; Raught, B.; Frappier, L. Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J. Virol. 2012, 86, 806–820. [Google Scholar] [CrossRef]
- Castillo, J.P.; Yurochko, A.D.; Kowalik, T.F. Role of human cytomegalovirus immediate-early proteins in cell growth control. J. Virol. 2000, 74, 8028–8037. [Google Scholar] [CrossRef]
- Wiebusch, L.; Hagemeier, C. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J. Virol. 1999, 73, 9274–9283. [Google Scholar]
- Kwon, Y.; Kim, M.N.; Young Choi, E.; Heon Kim, J.; Hwang, E.S.; Cha, C.Y. Inhibition of p53 transcriptional activity by human cytomegalovirus UL44. Microbiol. Immunol. 2012, 56, 324–331. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stewart, Z.A. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181–182, 475–481. [Google Scholar] [CrossRef]
- Margolis, M.J.; Pajovic, S.; Wong, E.L.; Wade, M.; Jupp, R.; Nelson, J.A.; Azizkhan, J.C. Interaction of the 72-kilodalton human cytomegalovirus IE1 gene product with E2F1 coincides with E2F-dependent activation of dihydrofolate reductase transcription. J. Virol. 1995, 69, 7759–7767. [Google Scholar]
- Castillo, J.P.; Frame, F.M.; Rogoff, H.A.; Pickering, M.T.; Yurochko, A.D.; Kowalik, T.F. Human cytomegalovirus IE1–72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J. Virol. 2005, 79, 11467–11475. [Google Scholar]
- Prichard, M.N.; Sztul, E.; Daily, S.L.; Perry, A.L.; Frederick, S.L.; Gill, R.B.; Hartline, C.B.; Streblow, D.N.; Varnum, S.M.; Smith, R.D.; et al. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 2008, 82, 5054–5067. [Google Scholar] [CrossRef]
- Zhang, Z.; Huong, S.M.; Wang, X.; Huang, D.Y.; Huang, E.S. Interactions between human cytomegalovirus IE1–72 and cellular p107: Functional domains and mechanisms of up-regulation of cyclin E/cdk2 kinase activity. J. Virol. 2003, 77, 12660–12670. [Google Scholar]
- Hagemeier, C.; Caswell, R.; Hayhurst, G.; Sinclair, J.; Kouzarides, T. Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J. 1994, 13, 2897–2903. [Google Scholar]
- Kalejta, R.F. Human cytomegalovirus pp71: A new viral tool to probe the mechanisms of cell cycle progression and oncogenesis controlled by the retinoblastoma family of tumor suppressors. J. Cell. Biochem. 2004, 93, 37–45. [Google Scholar] [CrossRef]
- Kalejta, R.F.; Shenk, T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. USA 2003, 100, 3263–3268. [Google Scholar] [CrossRef]
- Pajovic, S.; Wong, E.L.; Black, A.R.; Azizkhan, J.C. Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol. Cell. Biol. 1997, 17, 6459–6464. [Google Scholar]
- Pickering, M.T.; Kowalik, T.F. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 2006, 25, 746–755. [Google Scholar] [CrossRef]
- Johnson, R.A.; Yurochko, A.D.; Poma, E.E.; Zhu, L.; Huang, E.S. Domain mapping of the human cytomegalovirus IE1–72 and cellular p107 protein-protein interaction and the possible functional consequences. J. Gen. Virol. 1999, 80, 1293–1303. [Google Scholar]
- Ahn, J.H.; Hayward, G.S. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 1997, 71, 4599–4613. [Google Scholar]
- Wilkinson, G.W.; Kelly, C.; Sinclair, J.H.; Rickards, C. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 1998, 79, 1233–1245. [Google Scholar]
- Xiaofei, E.; Stadler, B.M.; Debatis, M.; Wang, S.; Lu, S.; Kowalik, T.F. RNA interference-mediated targeting of human cytomegalovirus immediate-early or early gene products inhibits viral replication with differential effects on cellular functions. J. Virol. 2012, 86, 5660–5673. [Google Scholar]
- Borden, K.L. Pondering the promyelocytic leukemia protein (PML) puzzle: Possible functions for PML nuclear bodies. Mol. Cell. Biol. 2002, 22, 5259–5269. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Promyelocytic leukemia-nuclear body proteins: Herpesvirus enemies, accomplices, or both? Future Viro. 2008, 3, 265–277. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef]
- Sommer, M.H.; Scully, A.L.; Spector, D.H. Transactivation by the human cytomegalovirus IE2 86-kilodalton protein requires a domain that binds to both the TATA box-binding protein and the retinoblastoma protein. J. Virol. 1994, 68, 6223–6231. [Google Scholar]
- Choi, K.S.; Kim, S.J.; Kim, S. The retinoblastoma gene product negatively regulates transcriptional activation mediated by the human cytomegalovirus IE2 protein. Virology 1995, 208, 450–456. [Google Scholar] [CrossRef]
- Speir, E.; Modali, R.; Huang, E.S.; Leon, M.B.; Shawl, F.; Finkel, T.; Epstein, S.E. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 1994, 265, 391–394. [Google Scholar]
- Tsai, H.L.; Kou, G.H.; Chen, S.C.; Wu, C.W.; Lin, Y.S. Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J. Biol. Chem. 1996, 271, 3534–3540. [Google Scholar]
- Bonin, L.R.; McDougall, J.K. Human cytomegalovirus IE2 86-kilodalton protein binds p53 but does not abrogate G1 checkpoint function. J. Virol. 1997, 71, 5861–5870. [Google Scholar]
- Song, Y.J.; Stinski, M.F. Effect of the human cytomegalovirus IE86 protein on expression of E2F-responsive genes: A DNA microarray analysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2836–2841. [Google Scholar] [CrossRef]
- Ruger, B.; Klages, S.; Walla, B.; Albrecht, J.; Fleckenstein, B.; Tomlinson, P.; Barrell, B. Primary structure and transcription of the genes coding for the two virion phosphoproteins pp65 and pp71 of human cytomegalovirus. J. Virol. 1987, 61, 446–453. [Google Scholar]
- Hensel, G.M.; Meyer, H.H.; Buchmann, I.; Pommerehne, D.; Schmolke, S.; Plachter, B.; Radsak, K.; Kern, H.F. Intracellular localization and expression of the human cytomegalovirus matrix phosphoprotein pp71 (ppUL82): Evidence for its translocation into the nucleus. J. Gen. Virol. 1996, 77, 3087–3097. [Google Scholar] [CrossRef]
- Baldick, C.J., Jr.; Marchini, A.; Patterson, C.E.; Shenk, T. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 1997, 71, 4400–4408. [Google Scholar]
- Rechter, S.; Scott, G.M.; Eickhoff, J.; Zielke, K.; Auerochs, S.; Muller, R.; Stamminger, T.; Rawlinson, W.D.; Marschall, M. Cyclin-dependent Kinases Phosphorylate the Cytomegalovirus RNA Export Protein pUL69 and Modulate Its Nuclear Localization and Activity. J. Biol. Chem. 2009, 284, 8605–8613. [Google Scholar] [CrossRef]
- Marschall, M.; Freitag, M.; Suchy, P.; Romaker, D.; Kupfer, R.; Hanke, M.; Stamminger, T. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 2003, 311, 60–71. [Google Scholar] [CrossRef]
- Schregel, V.; Auerochs, S.; Jochmann, R.; Maurer, K.; Stamminger, T.; Marschall, M. Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Gen. Virol. 2007, 88, 395–404. [Google Scholar] [CrossRef]
- Michel, D.; Kramer, S.; Hohn, S.; Schaarschmidt, P.; Wunderlich, K.; Mertens, T. Amino acids of conserved kinase motifs of cytomegalovirus protein UL97 are essential for autophosphorylation. J. Virol. 1999, 73, 8898–8901. [Google Scholar]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef]
- Winkler, M.; Stamminger, T. A specific subform of the human cytomegalovirus transactivator protein pUL69 is contained within the tegument of virus particles. J. Virol. 1996, 70, 8984–8987. [Google Scholar]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006, 80, 8390–8401. [Google Scholar] [CrossRef]
- E, X.; Savidis, G.; Chin, C.R.; Wang, S.; Lu, S.; Brass, A.L.; Kowalik, T.F. A Novel DDB2-ATM Feedback Loop Regulates Human Cytomegalovirus Replication. J. Virol. 2014, 88, 2279–2290. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Orazio, N.I.; Naeger, C.M.; Karlseder, J.; Weitzman, M.D. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 2011, 85, 1887–1892. [Google Scholar] [CrossRef]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi’s Sarcoma-Associated Herpesvirus Induces the ATM and H2AX DNA Damage Response Early during De Novo Infection of Primary Endothelial Cells, Which Play Roles in Latency Establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef]
- Chang, L.; Godinez, W.J.; Kim, I.H.; Tektonidis, M.; de Lanerolle, P.; Eils, R.; Rohr, K.; Knipe, D.M. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. USA 2011, 108, E136–E144. [Google Scholar] [CrossRef]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar]
- Wilkinson, D.E.; Weller, S.K. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 2003, 55, 451–458. [Google Scholar] [CrossRef]
- Kaufer, B.B.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 2011, 208, 605–615. [Google Scholar] [CrossRef]
- Li, M.; Mizuuchi, M.; Burke, T.R., Jr.; Craigie, R. Retroviral DNA integration: Reaction pathway and critical intermediates. EMBO J. 2006, 25, 1295–1304. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Carson, C.T.; Schwartz, R.A.; Lilley, C.E. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 2004, 3, 1165–1173. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Dell’Aquila, M.L.; Spector, D.H. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2000, 97, 853–858. [Google Scholar] [CrossRef]
- Kwak, S.; E, X.; Kowalik, T.F. An HCMV virion-associated tegument protein initiates activation of the host DNA damage response to infection. University of Massachusetts Medical School: Worcester, MA, USA, 2014; to be submitted for publication. [Google Scholar]
- Millhouse, S.; Su, Y.H.; Zhang, X.; Wang, X.; Song, B.P.; Zhu, L.; Oppenheim, E.; Fraser, N.W.; Block, T.M. Evidence that herpes simplex virus DNA derived from quiescently infected cells in vitro, and latently infected cells in vivo, is physically damaged. J. Neurovirol. 2010, 16, 384–398. [Google Scholar] [CrossRef]
- Stich, H.F.; Hsu, T.C.; Rapp, F. Viruses and Mammalian Chromosomes. I. Localization of Chromosome Aberrations after Infection with Herpes Simplex Virus. Virology 1964, 22, 439–445. [Google Scholar] [CrossRef]
- Peat, D.S.; Stanley, M.A. Chromosome damage induced by herpes simplex virus type 1 in early infection. J. Gen. Virol. 1986, 67, 2273–2277. [Google Scholar] [CrossRef]
- Schramayr, S.; Caporossi, D.; Mak, I.; Jelinek, T.; Bacchetti, S. Chromosomal damage induced by human adenovirus type 12 requires expression of the E1B 55-kilodalton viral protein. J. Virol. 1990, 64, 2090–2095. [Google Scholar]
- RW, H.Y.; Oakes, J.E.; Kudler, L. In vitro repair of the preexisting nicks and gaps in herpes simplex virus DNA. Virology 1977, 76, 286–294. [Google Scholar] [CrossRef]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J. Virol. 2011, 85, 12585–12593. [Google Scholar]
- Soutoglou, E.; Misteli, T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008, 320, 1507–1510. [Google Scholar] [CrossRef]
- McVoy, M.A.; Adler, S.P. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J. Virol. 1994, 68, 1040–1051. [Google Scholar]
- E, X.; Kowalik, T.F. A role for host DNA polymerases in the replication of herpesviral DNA. University of Massachusetts Medical School: Worcester, MA, USA, 2014; to be submitted for publication. [Google Scholar]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Cuchet-Lourenco, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 2012, 86, 11209–11222. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
E, X.; Kowalik, T.F. The DNA Damage Response Induced by Infection with Human Cytomegalovirus and Other Viruses. Viruses 2014, 6, 2155-2185. https://doi.org/10.3390/v6052155
E X, Kowalik TF. The DNA Damage Response Induced by Infection with Human Cytomegalovirus and Other Viruses. Viruses. 2014; 6(5):2155-2185. https://doi.org/10.3390/v6052155
Chicago/Turabian StyleE, Xiaofei, and Timothy F. Kowalik. 2014. "The DNA Damage Response Induced by Infection with Human Cytomegalovirus and Other Viruses" Viruses 6, no. 5: 2155-2185. https://doi.org/10.3390/v6052155
APA StyleE, X., & Kowalik, T. F. (2014). The DNA Damage Response Induced by Infection with Human Cytomegalovirus and Other Viruses. Viruses, 6(5), 2155-2185. https://doi.org/10.3390/v6052155