1. Introduction
Gene delivery is one of the most fundamental techniques of molecular biology, and a technological basis for
in vitro and
in vivo gene therapy. By definition, gene transfection involves the delivery of nucleic acids (DNA or RNA) into cells to genetically modify them. Expression of transgenes in cell cultures creates a suitable system to determine the regulation and function of a desired gene, and in turn the function of proteins and their network systems. Additionally, transfection has revolutionized scientific industries allowing for the development of large-scale recombinant protein production including antibodies, vaccines and viral vectors [
1,
2].
Methods developed for transfection can broadly be classified into three categories; biological, chemical and physical [
3]. The choice of method depends heavily on the type of system to be transfected, the size of the transgene and whether the resulting output requires transient or stable transgene expression. To achieve successful gene transfer, nucleic acids have to overcome several cellular barriers including surface adsorption and entry, degradation during intracellular trafficking and finally be able to induce transgene expression within the nucleus. Traditionally, transfection of cell cultures is achieved by the use of chemical transfection reagents, which deliver the nucleic acids into cells. Calcium phosphate was the first transfection reagent to be developed and works on the basis that positively charged calcium ions bind to the negatively charged phosphate backbone of DNA and form a co-precipitation complex for transportation into cells through endocytosis [
4]. Since calcium phosphate, many different transfection reagents have been developed including cationic lipids (most popular), polycationic polymers [
5], and cationic amino acids [
6]. All these transfection reagents work on the same basic principal in that the positively charged chemicals interact and condense the negatively charged DNA to form positively charged complexes for easy transport through the negative cell membranes. A successful transfection reagent should have minimal cytotoxicity, high transfection efficiency, be easy to reproduce and be inexpensive, particularly for large-scale transfection processes in industry. However, as with every technology there are limitations; transfection reagents have low efficacy, can be expensive and it is difficult to target them to specific cell types.
On the other hand, viral vectors have also been developed for gene delivery purposes within laboratory research but are mainly used
in vivo for gene therapy applications. The most successful viral vectors to date include adenovirus, lentivirus and adeno-associated virus [
7,
8,
9,
10,
11]. While these vectors are superior in their gene delivery efficacy compared with non-viral vectors, they have various limitations. Firstly, they can have a limited packing capacity, which restricts the size of the transgene that can be engineered into their genome. Secondly, they have a complex protein structure, which makes their production complicated, less efficient and very expensive. Finally, they are not deemed safe for applications such as production of recombinant proteins for human purposes as they have a broad tropism for mammalian cells.
Bacteriophages (phage), viruses that infect only bacteria, are attracting increasing attention as promising new biomaterials in the field of gene delivery. Mainly, filamentous M13 bacteriophages are being developed as a new type of vectors for safe and targeted systemic administration of transgenes for
in vivo applications [
12,
13,
14,
15]. They have a number of advantages over the use of traditional viral and non-viral vectors; firstly, their protein coat consists of a repeating protein unit arranged in an alpha helical array. This bestows the phage with an unlimited DNA packaging capacity, as the capsid coat merely needs to elongate to accommodate the transgene. Therefore, the bacteriophage is efficient at condensing and packing the DNA. Secondly, the protein coat has a high tolerance for mutations, allowing peptide ligands to be easily introduced to achieve ligand-directed transduction of the desired cell type. Additionally, they are safe as they have long been administered to humans for the treatment of bacterial infections and have been approved by the Food and Drug Administration (FDA-USA) for use in food preparations [
16,
17]. Lastly, phage vectors are easy to produce at high titers and at low costs, which is highly desirable for large scale industrial processes. Previously, we reported the development of an M13 phage-based vector consisting of a mammalian transgene cassette flanked by inverted terminal repeats (ITR) from adeno-associated virus serotype 2 (AAV2), incorporated in an intergenomic region of the phage genome. Because bacteriophages have no tropism for mammalian cells, and are therefore unable to enter and transduce these cells, we genetically engineered the phage to display the double cyclic RGD4C (CDCRGDCFC) ligand, on the phage capsid, to bind to αv integrin receptors on the cell surface to allow phage internalization and subsequently expression of the gene of interest. This vector, named AAV/phage or AAVP, has shown promise for specific tumor targeting
in vitro and
in vivo [
14]. However, bacteriophages remain poor delivery vectors in comparison to traditional viral vectors, as they have no intrinsic strategies for mammalian cell transduction. Therefore, strategies need to be explored to allow efficient phage binding, entry into mammalian cells and subsequent increase of their gene transfer ability, which should subsequently develop their use in a wide range of gene transfer applications.
Combination of viral vectors with traditional transfection reagents has previously been reported for the development of
in vivo gene delivery vectors [
18,
19]. Here, we report a new hybrid bacteriophage-based system for general gene delivery processes. We have taken two of the central themes of gene transfer, biological (phage) and chemical (transfection reagents), and combined them into a superior gene delivery platform (
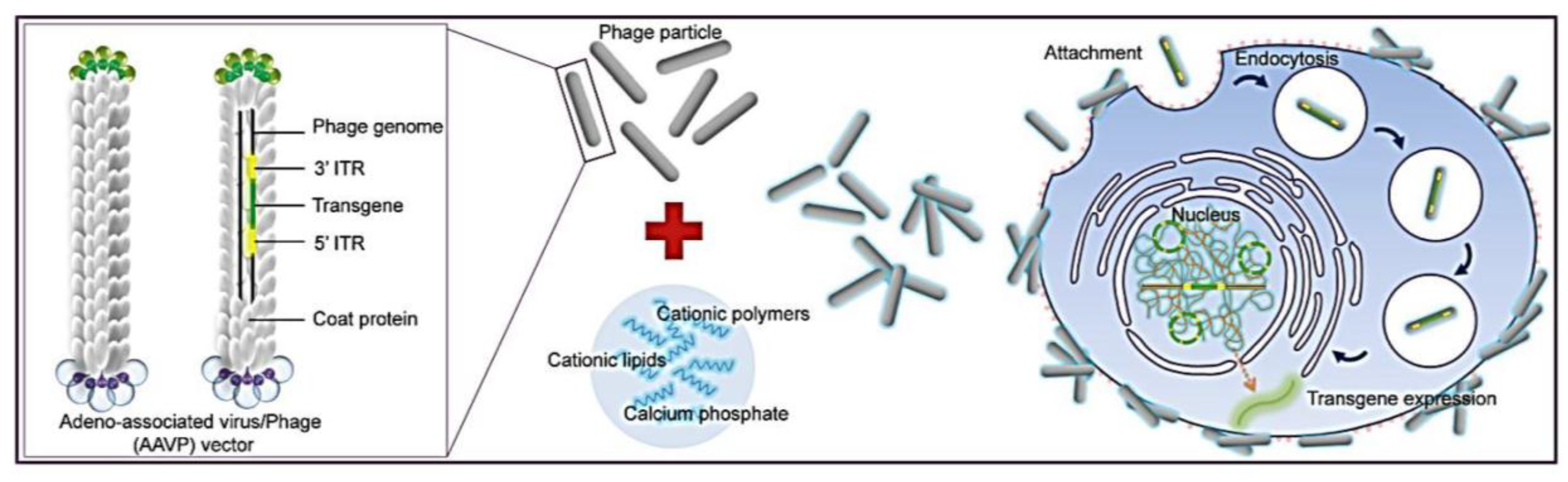
Figure 1). To allow phage entry into mammalian cells to achieve gene delivery, we evaluated the bacteriophage vector system in combination with chemical modifications of the phage capsid using transduction-enhancing agents. Three different types of transduction reagents were tested including cationic lipids, polycationic polymers and calcium phosphate (
Figure 1). We established optimal transduction conditions for the phage-reagent vector system and investigated the mechanisms by which increased gene delivery efficacy was achieved. Integration of phage with traditional transfection reagents resulted in increased ability of phage binding to the cell surfaces, entry into cells and subsequent efficient gene expression by the phage vectors. Moreover, we determined that gene delivery by the hybrid vector, consisting of cationic polymers, was highly efficient in stable cell line generation. Finally, importantly, we demonstrated that integration of both chemical modification and genetic engineering of the phage capsid through display of the RGD4C ligand resulted in further increase of phage-mediated gene delivery compared to each modification alone. Here, we propose that these hybrid phage complexes have the potential to be an industry wide tool for high gene delivery output as well as applications within laboratory research.
Figure 1.
Schematic diagram of the hybrid bacteriophage/transfection reagents system. Negatively charged bacteriophage vectors were electrostatically mixed with transfection reagents to form hybrid bacteriophage complexes. A hybrid vector consists of the filamentous M13 bacteriophage and a transfection reagent. The phage serves as a transgene carrier. ITR, inverted terminal repeats of adeno-associated virus serotype 2 (AAV2).
Figure 1.
Schematic diagram of the hybrid bacteriophage/transfection reagents system. Negatively charged bacteriophage vectors were electrostatically mixed with transfection reagents to form hybrid bacteriophage complexes. A hybrid vector consists of the filamentous M13 bacteriophage and a transfection reagent. The phage serves as a transgene carrier. ITR, inverted terminal repeats of adeno-associated virus serotype 2 (AAV2).
2. Materials and Methods
2.1. Construction and Production of Bacteriophage Vectors
As phage vectors, we used the previously reported AAVP vector. To construct these vectors, phage was genetically manipulated to carry a mammalian transgene cassette encoding the cytomegalovirus (CMV) promoter-driven transgene, flanked by inverted terminal repeats (ITR)s from AAV2. Some phage vectors were also further manipulated for ligand-mediated gene delivery by incorporating copies of the RGD4C tumor targeting peptide on the pIII minor coat protein of the phage, in order to generate the RGD4C-phage. Phage vectors, with RGD4C (RGD4C-phage) or without RGD4C (phage), were amplified, isolated and purified from the culture supernatant of host bacteria (
Escherichia coli K91) as previously described [
13]. Phage particles were sterile filtered through 0.45 µm filters, then titrated using a qNano analyzer (IZON Science Ltd. T., Oxford, UK) based on a coulter technique also known as resistive pulse sensing, and expressed as mg/mL.
2.2. Chemical Modification of Phage Vectors
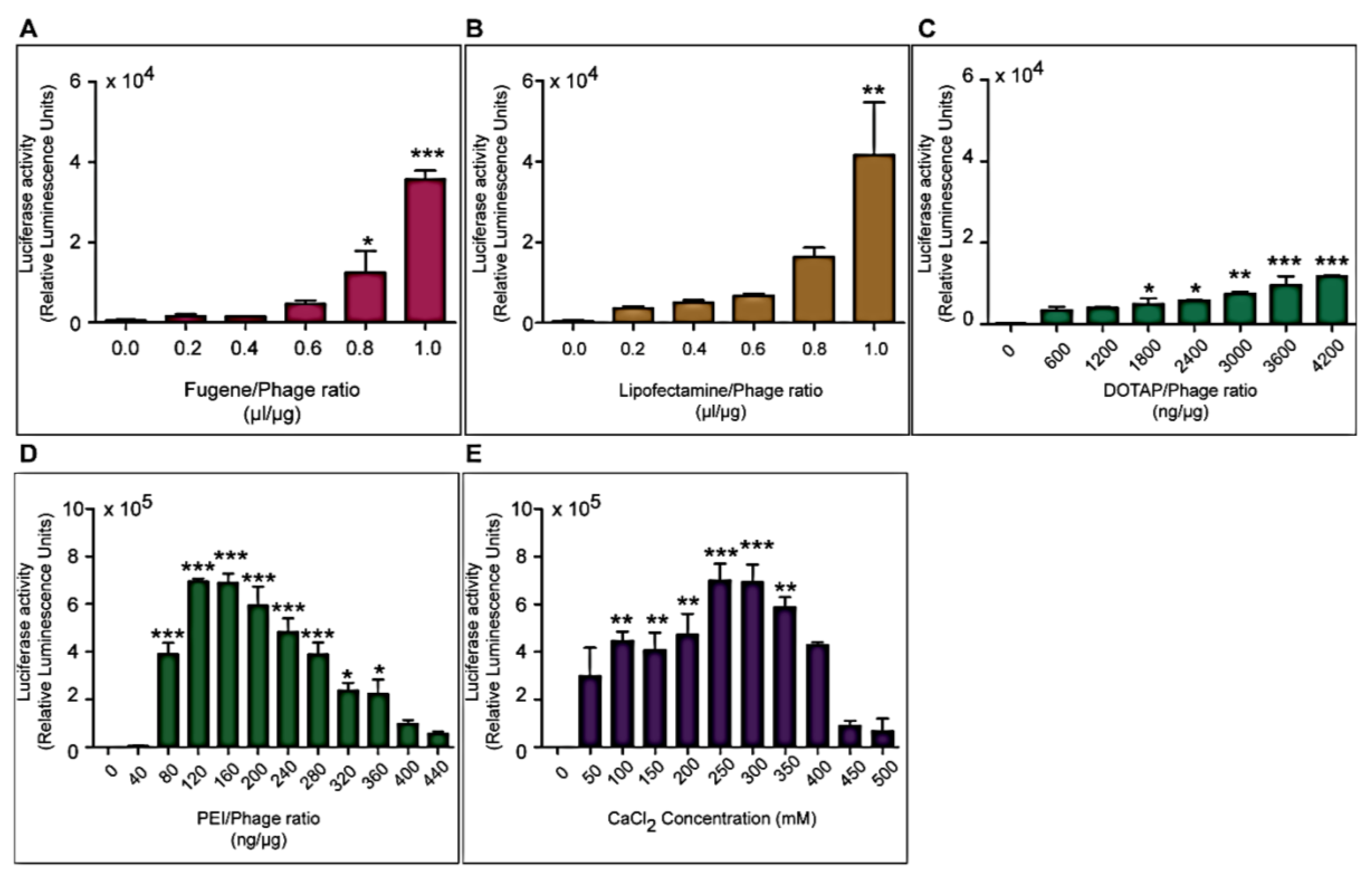
Three different types of transfection reagents were tested in combination with the phage vectors including cationic lipids, cationic polymers and calcium phosphate (CaPi). For cationic lipids and polymers, desired concentrations of chemicals were added to 25 μg of phage vector preparations in complete Dulbecco’s Modified eagle’s Medium (DMEM, Sigma, Dorset, UK). Solutions were incubated for 15 min at room temperature to allow for the formation of complexes before initiation of cell transduction. For calcium phosphate, the desired concentration of CaCl2 was added to 25 μg phage vector prepared in double distilled water. An equal volume of 2× Hepes-buffered saline solution (HBS) was added to allow the formation of precipitates.
2.3. Cell Culture
Human Embryonic Kidney (HEK293) cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The human MCF-7 breast cancer cell line was from the Cancer Research UK (London, UK)). Cells were cultured in DMEM, supplemented with 10% Fetal Bovine Serum (FBS, Sigma), Penicillin (100 units/mL, Sigma), Streptomycin (100 μg/mL, Sigma) and l-Glutamine (2 mmol/mL, Sigma). Cells were maintained in a humidified atmosphere of 37 °C and 5% CO2 and passaged every 3–4 days once they reach 70%–80% confluence.
2.4. In Vitro Cell Transduction by Phage-Derived Vector Complexes
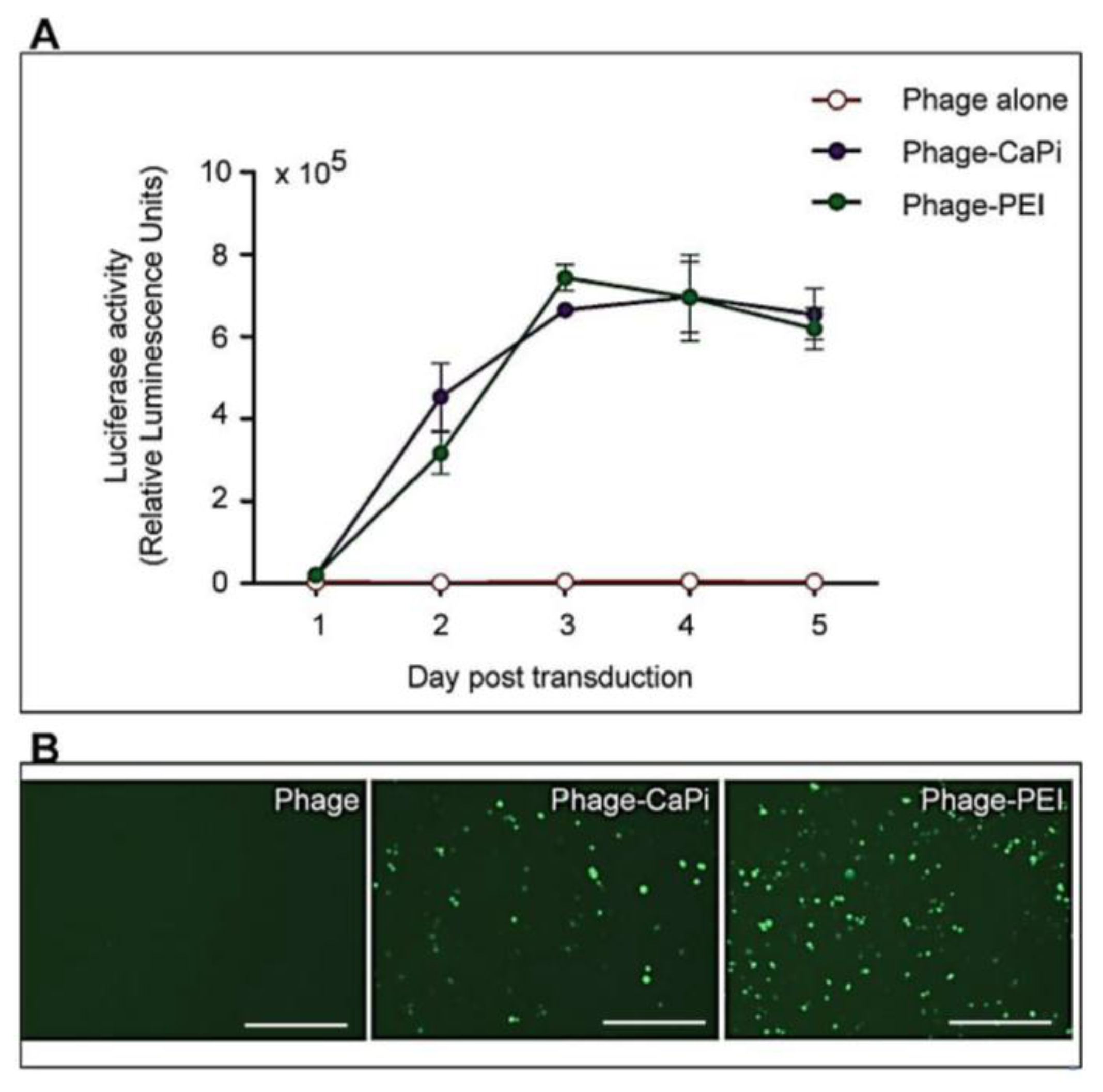
Cells were seeded at 3 × 104 cells/well in 48-well plates and allowed to proliferate until 60%–80% confluent. Complexes of phage vectors and transfection reagents prepared at optimal ratios in serum-free media, or control phage vector alone were applied to the cells, followed by incubation at 37 °C for 4 h. Next complete media, containing FBS, was administered and the cells were incubated at 37 °C to allow for transgene expression. Transduction efficiency of the phage complexes was determined by using phage carrying the green fluorescent protein (GFP) or firefly luciferase (Luc) reporter genes. GFP expression was evaluated using a Nikon eclipse TE200-S fluorescent microscope (Nikon, Surrey, UK). Luc reporter gene expression in transduced cells was determined by using the Promega Steady-Glo® Luciferase Assay kit following the manufacturer’s protocol and quantified using a Promega plate reader (Promega, Southampton, UK). Data were normalized to 100 µg protein levels as determined by the Bradford assay and presented as relative luminescence units per 100 µg protein. Cell viability was analyzed by CellTiter-glo® cell viability assay kit; following the manufacturer’s protocol and quantified using a Promega plate reader. All cell transduction experiments were repeated three times and performed in triplicates.
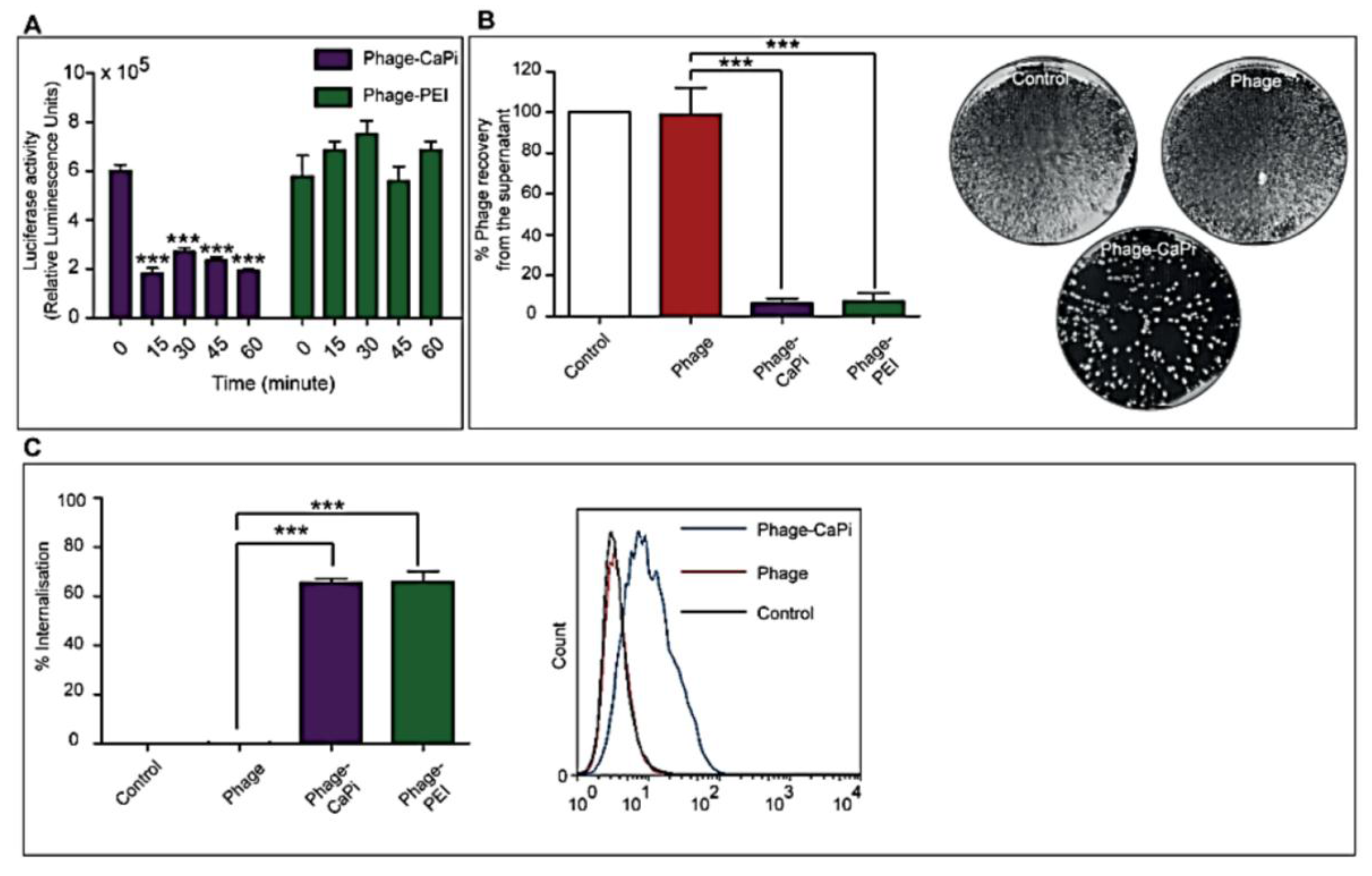
2.5. In Vitro Depletion Assay
HEK293 cells in 48-well plates, at 70%–80% confluence, were treated with phage vectors prepared at optimal ratios with the desired transfection reagents. The plates were placed on ice for 1 h to prevent internalization followed by collection of supernatants, which were then subjected to serial dilution in 1× phosphate buffered saline (PBS) solution. Experiments were performed in triplicates, repeated twice, and K91 Kan bacterial infection method was used to quantify the number of phage vector particles by counting transducing units as previously reported [
13].
2.6. Internalization Assay
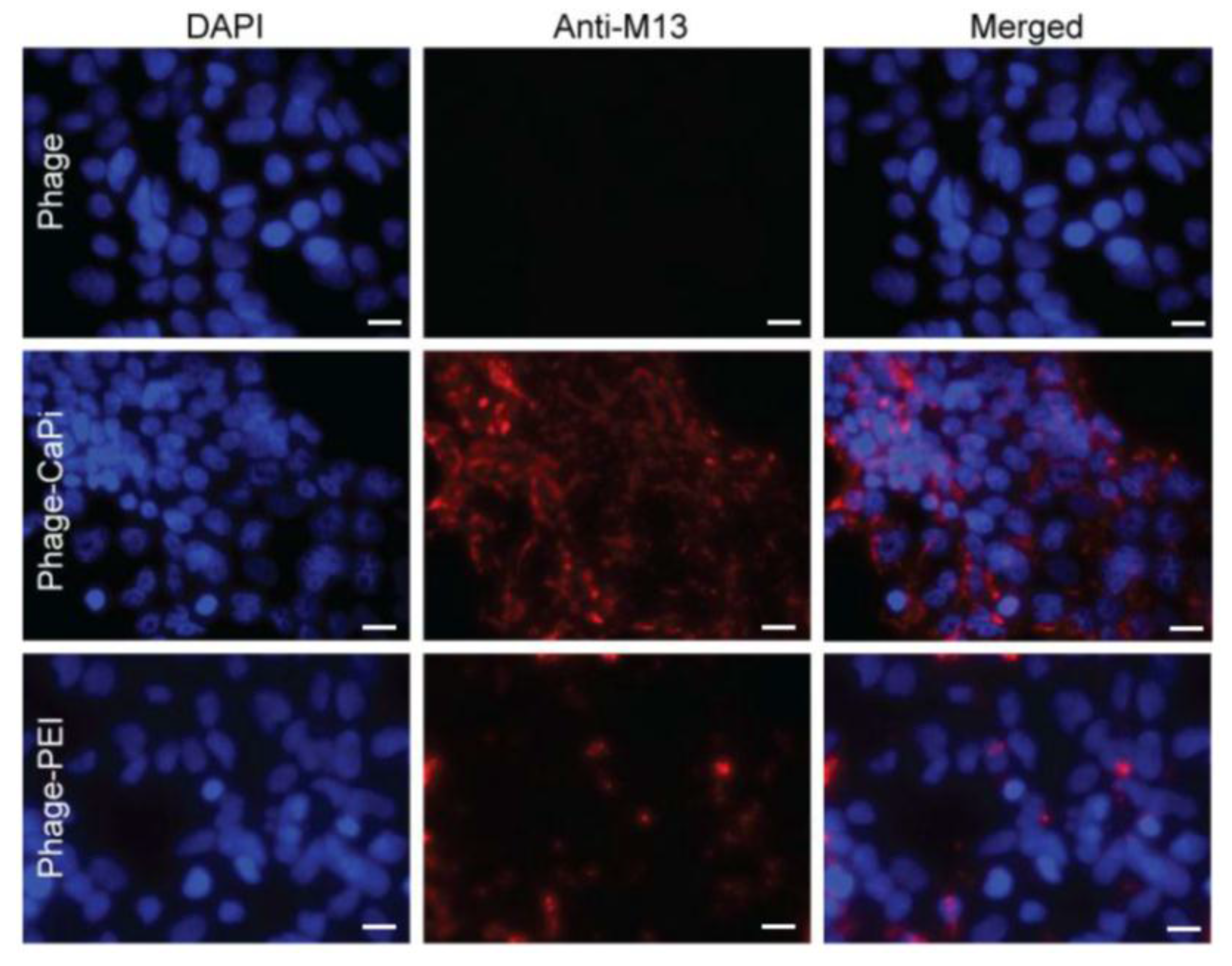
Phage vector particles internalized in HEK293 cells were quantified as previously reported [
20]. Briefly, unbound and surface bound vector particles were removed from cells by washing with 1× PBS and trypsin, respectively. Cells were centrifuged at 200 rpm for 5 min and fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature. Cells were blocked in 0.1% saponin in 2% bovine albumin serum (BSA)-PBS for 30 min. To detect internalized phage, cells were stained with a rabbit anti-M13 phage antibody (dilution 1:1000) in 0.1% saponin in 1% BSA-PBS for 1 h at room temperature. Cells were centrifuged and resuspended in 0.1% saponin in 1% BSA-PBS followed by incubation with a goat anti-rabbit AlexaFluor-647 (dilution 1:500) for 1 h at room temperature. For analysis, cells were washed in 0.1% saponin-PBS and resuspended in PBS. Fluorescence-activated cell sorting analysis was carried out using a FACSCalibur Flow cytometer (BD Biosciences, Oxford, UK) equipped with an argon-ion laser (488 nm) and red-diode laser (365 nm). Experiments were repeated twice, and 10,000 gated cells per triplicate wells were used as the mean fluorescence intensity. Results were analyzed using Flowjo software (TreeStar Ashland, OR, USA).
2.7. Generation of Stable Cell Lines
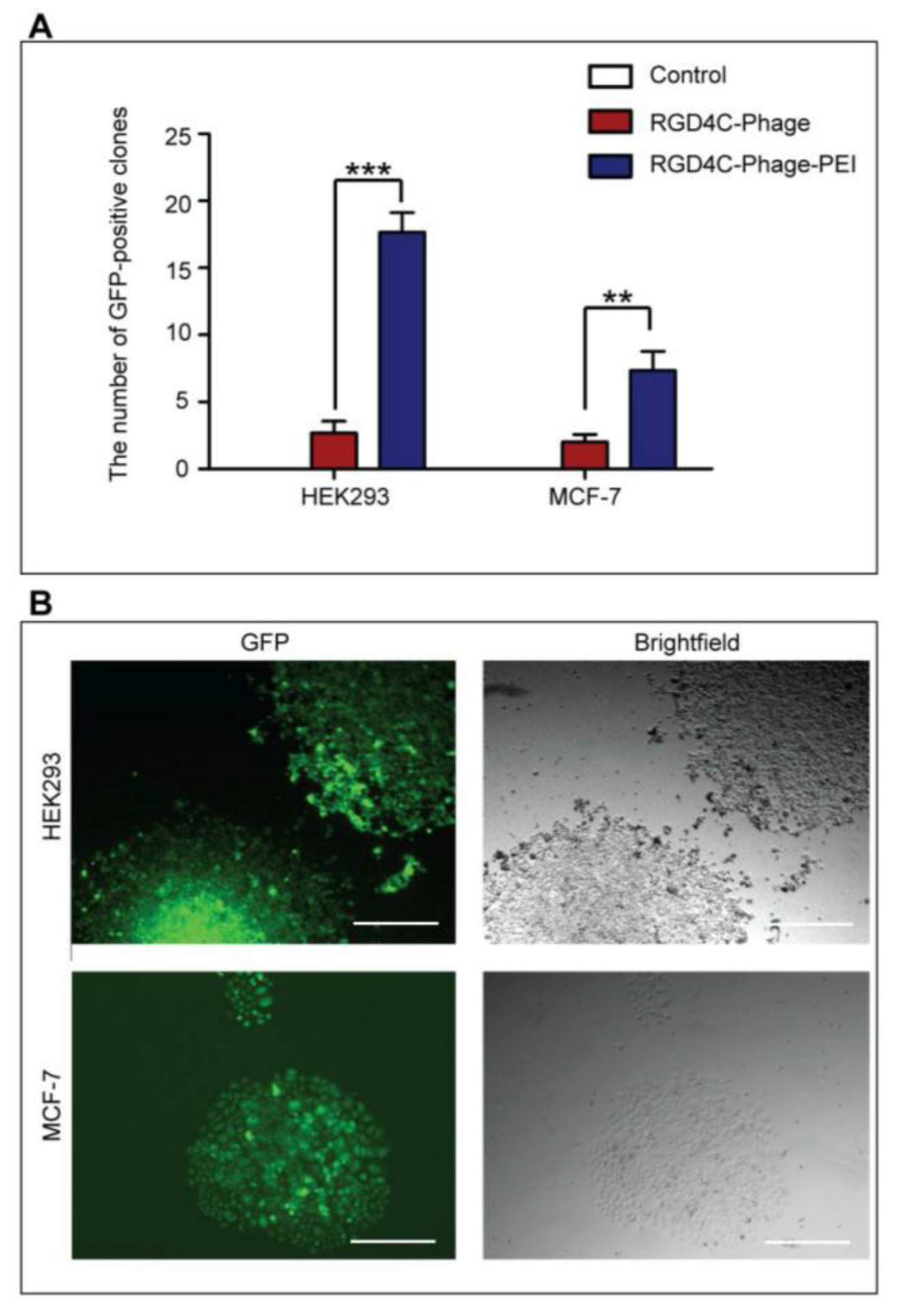
HEK293 and MCF-7 monolayer cell cultures in 12-well plates were treated with phage vectors carrying GFP and a puromycin-resistant gene (puror), combined or not with transfection reagents. At day 3 post-transduction, cells were trypsinised and resuspended in DMEM containing 1 μg/mL puromycin antibiotic, which was replenished every 3 days. After two weeks, puromycin-resistant clones were visible via fluorescent microscope and were subsequently pooled and passaged to generate a population of stably transduced cells.
2.8. Confocal Microscopy
Cells were seeded on 18 mm2 coverslips in 12-well plates. Cells were incubated with phage vectors (prepared at optimal ratios with transfection reagents) for 4 h, washed with PBS and fixed in 4% PFA in PBS for 15 min at room temperature and quenched with 50 mmol/L ammonium chloride. Cells were permeabilized with 0.2% Triton X-100, washed and blocked with 2% BSA-PBS for 30 min. For phage staining, cells were incubated for 1 h with rabbit anti-M13 phage (1:1000) followed by secondary AlexaFluor-conjugated antibodies (dilution 1:750) with/without DAPI (dilution 1:2000) for 1 h at room temperature. Finally, cells were mounted in Mowiol mounting medium and images were acquired with a Leica SP5 confocal microscope (Leica, Milton Keynes, UK). Experiments were performed in triplicates and repeated twice.
2.9. Statistical Analysis
GraphPad Prism software (version 5.0, GraphPad Software, Inc., La Jolla CA, USA) was used to perform statistical analyses Error bars represent standard error of the mean (s.e.m). p values were generated by ANOVA (Analysis of Variance) and denoted as follows: *p < 0.05, **p <0.01, ***p < 0.001 and n.s., non-significant.
4. Discussion
The delivery of nucleic acids into mammalian cells through non-viral methods has origins as far back as the 1960s [
26]. The ultimate goal of transfection is to deliver nucleic acids into cells so as to investigate gene function. This goal can be accomplished by expression of exogenous genes. Manipulation of gene expression is a core technique in research areas such as drug development, cancer research, gene therapy, and tissue engineering [
27,
28]. Additionally,
in vivo gene therapy applications have created a need to develop safer and more efficient techniques for delivering nucleic acids to different organs and tissues [
8]. This
in vivo research requires proof of concept, which is made possible through
in vitro transfection experiments.
Calcium phosphate represents the oldest and most inexpensive chemical method for transfecting nucleic acids [
4]. Despite the simple and cost-effective nature of the calcium phosphate method, it is ineffective for hard-to-transfect cells, is very sensitive to changes in pH, often lacks reproducibility, and requires large quantities of DNA. Other gene delivery methods that were developed after the introduction of calcium phosphate methods include direct delivery via injection into the cell nucleus (microinjection), use of viral vectors, electrical currents and lipid mediated techniques. Although microinjection provides a direct method for delivery of nucleic acid for cells that are difficult to transfect, it is low-throughput and a technique that is difficult to master. Electroporation has also been introduced and relies on an electrical field to transiently increase cell permeability; however, the application of an electrical field causes substantial cytotoxicity.
An alternative and more efficient method for delivering nucleic acid into cells is the use of viral vectors [
3]. The use of viral vectors is a common methodology for efficiently delivering nucleic acid, especially in hard-to-transfect cells. Generation of recombinant viruses requires the packaging of exogenous DNA within the viral genome and subsequent delivery through infection of the target cell. The use of viral vectors was used as early as the late 1970s to express functional mRNA and protein. Although viral transduction is an efficient and effective option, some disadvantages include viral recombination, off-target effects, immune response induction, and possible oncogenic effects [
29].
In brief, gene delivery continues to play a major role in a wide range of applications; however, the disadvantages of current gene transfer strategies necessitate more robust methods for nucleic acid delivery. Bacteriophage which has been recently described as a promising new generation of gene delivery vectors to mammalian has faced limited progress. Indeed, phage has no tropism for mammalian cells and thus cannot enter these cells to deliver gene expression. Genetic engineering of the phage has been shown to mediate gene delivery by display of ligands on the phage capsid to allow binding to a mammalian receptor and subsequent internalization of phage in mammalian cells. Herein, we show that chemical modification of the phage capsid allows phage entry into mammalian cells and subsequent expression of the gene of interest. Importantly, combination of both genetic and chemical modifications of the phage capsid further enhances gene delivery compared to each individual approach. Actually, we have developed a simple and effective method to enhance transduction efficiency of bacteriophage-based vectors using a combination of both conventional transfection reagents and ligand-directed transduction. Chemical modification has been combined with eukaryotic viral vectors to transfer genes to mammalian cells [
30,
31]. Genetic modification has also been applied to animal viral vectors to improve specificity; but to our knowledge, this is the first report showing combination of these two modifications into one single gene delivery particle. Moreover, there have been no reports regarding the application of filamentous bacteriophage complex with CaPi for gene transfer. The system we have described here has a number of advantages, and the results are encouraging.
In conclusion, we have presented a novel strategy to advance phage-based gene transfer by combining phage with conventional transfection reagents. Importantly, we report that integration of chemical modification with ligand-directed phage transduction further enhances phage-mediated gene delivery. This proof-of-concept study successfully shows that bacteriophage-mediated gene transfer into mammalian cells can be improved. Future studies to assess efficacy of these phage complexes in additional cell lines and applications, should provide further characterization of these hybrid phage complexes.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






