
In Search of Pathogens: Transcriptome-Based Identification of Viral Sequences from the Pine Processionary Moth (Thaumetopoea pityocampa)

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
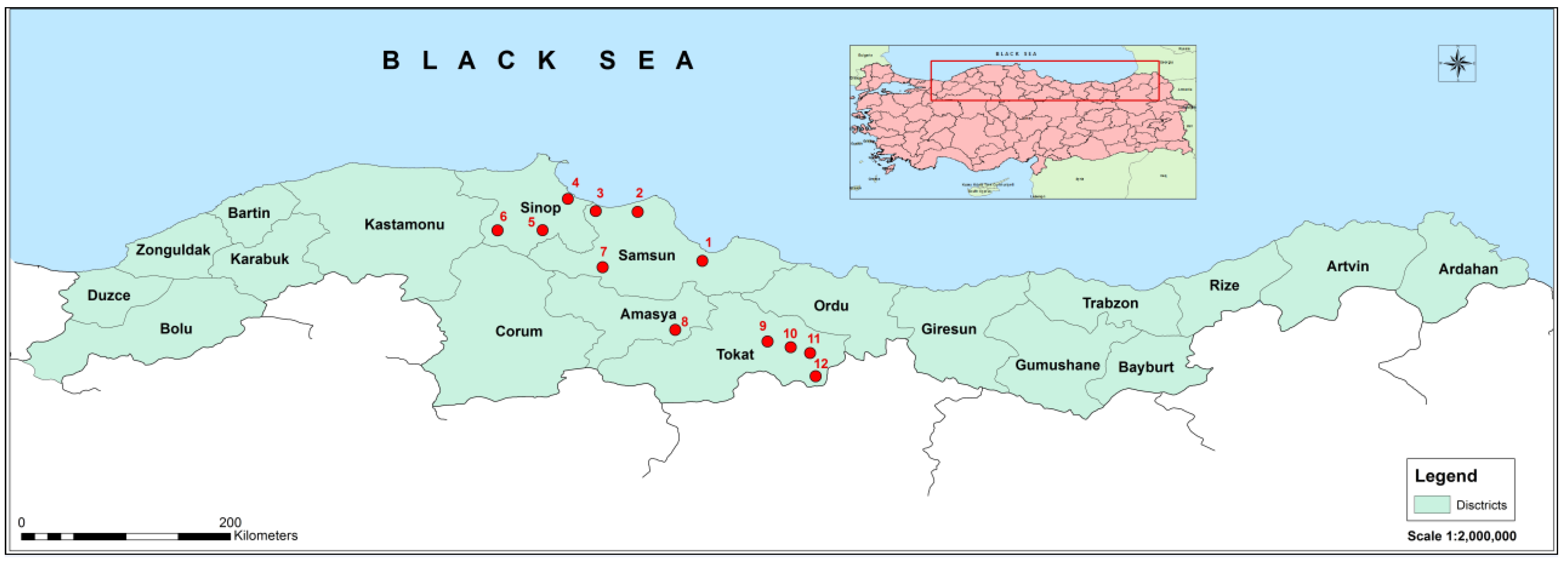
2.1. Sampling and RNA Isolation

2.2. Next Generation Sequencing and Raw Data Processing
2.3. De Novo Transcriptome Assembly
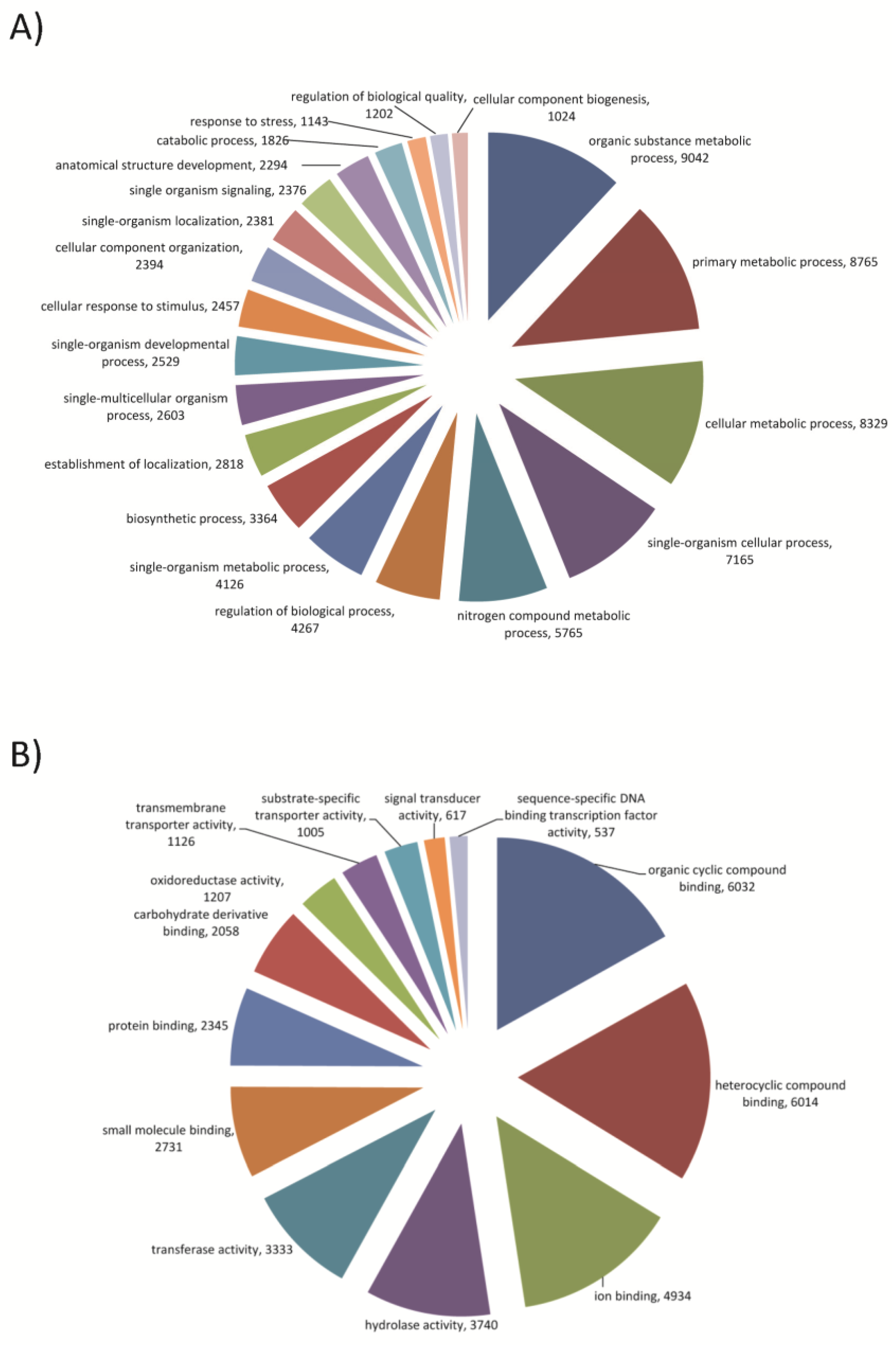
2.4. Functional Annotations and GO Terms Assignment
2.5. Virus Sequences Detection, Identification and Phylogenetic Analyses
2.6. RNAseq Expression Analysis and Tissue Specific Transcripts Identification
3. Results and Discussion
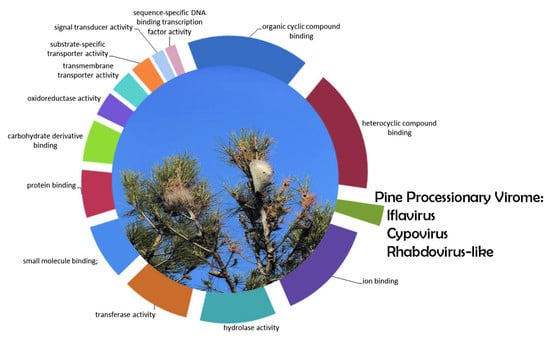
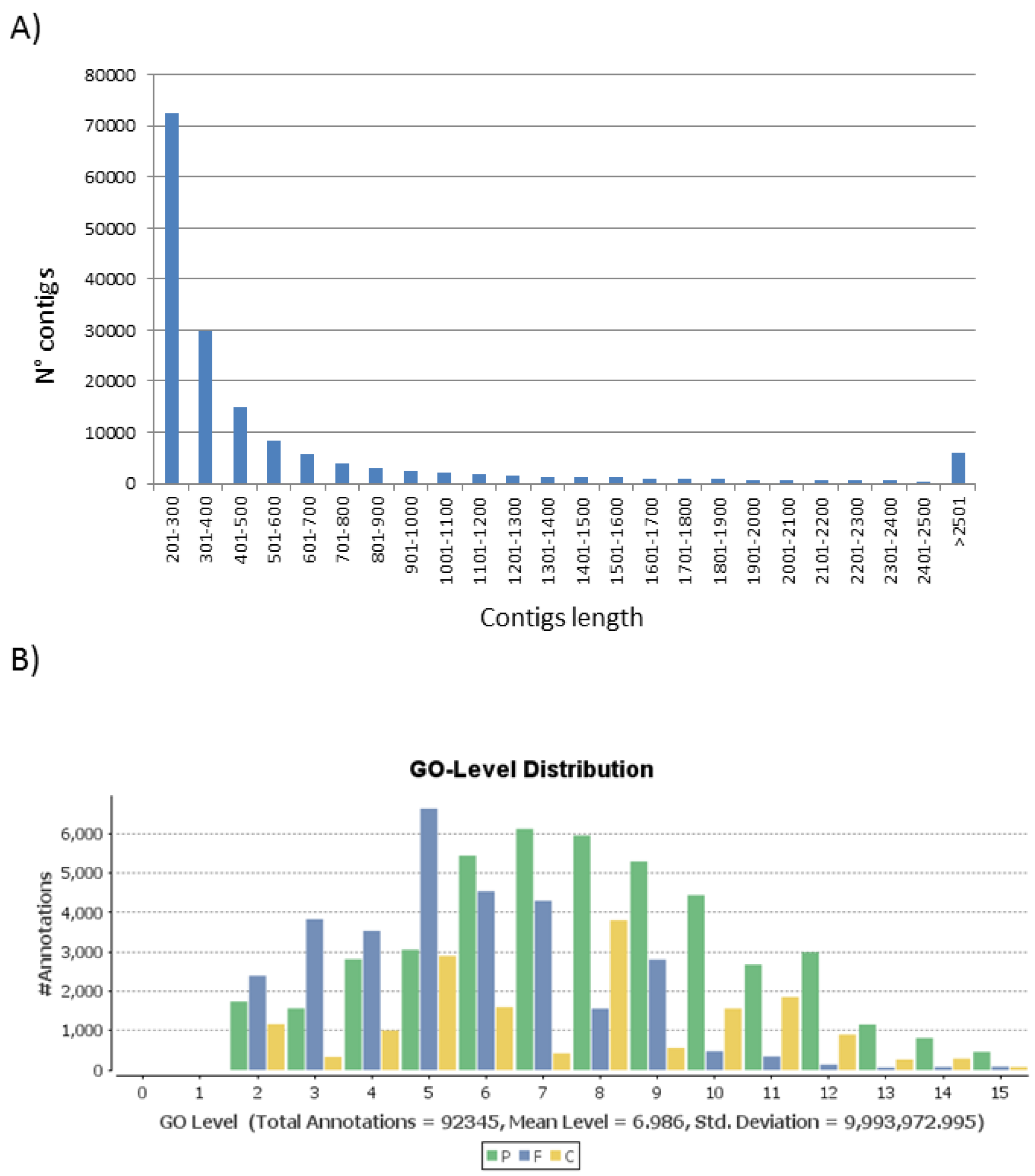
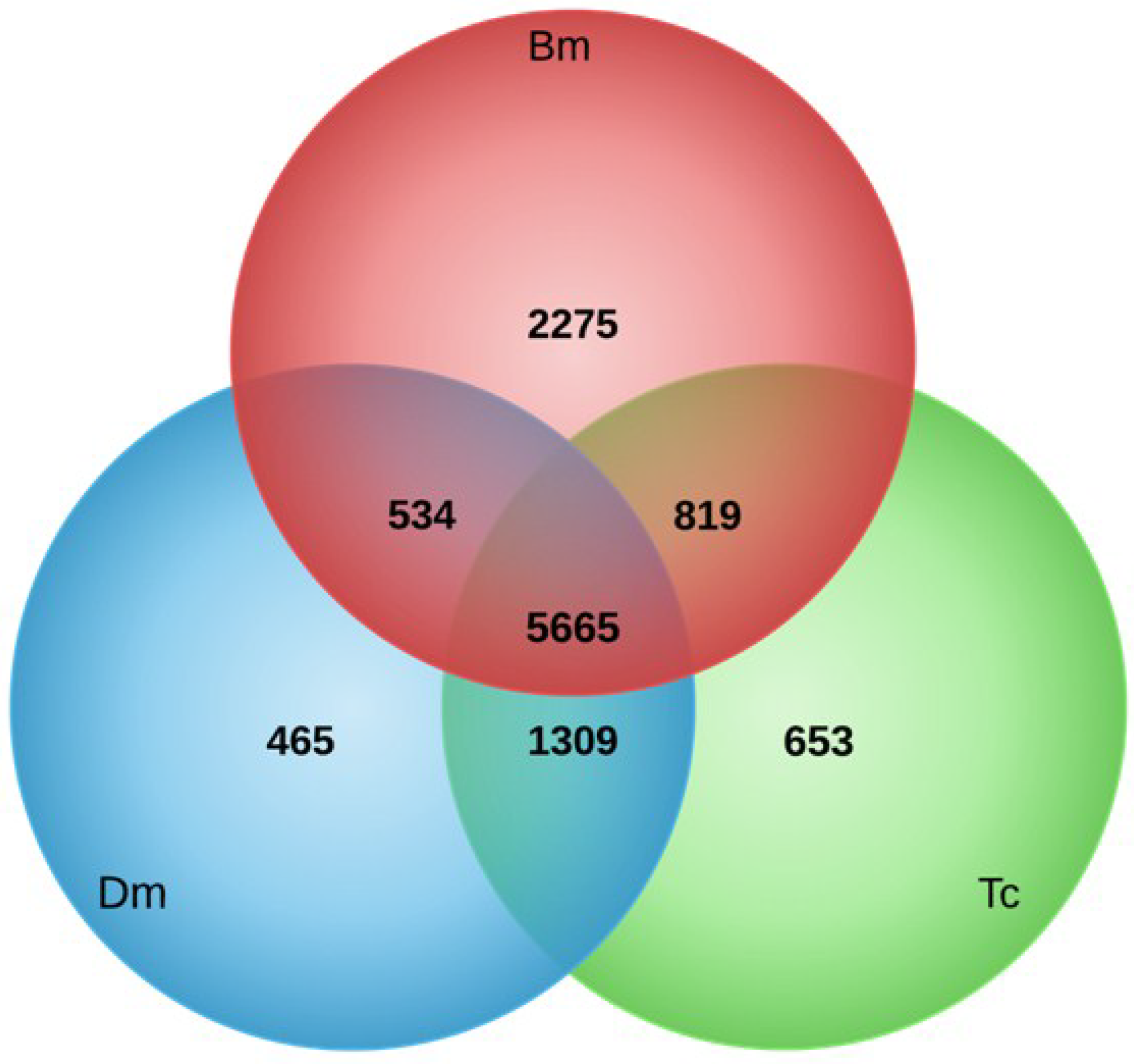
3.1. T. pityocampa Transcriptome Assembly and Functional Annotation

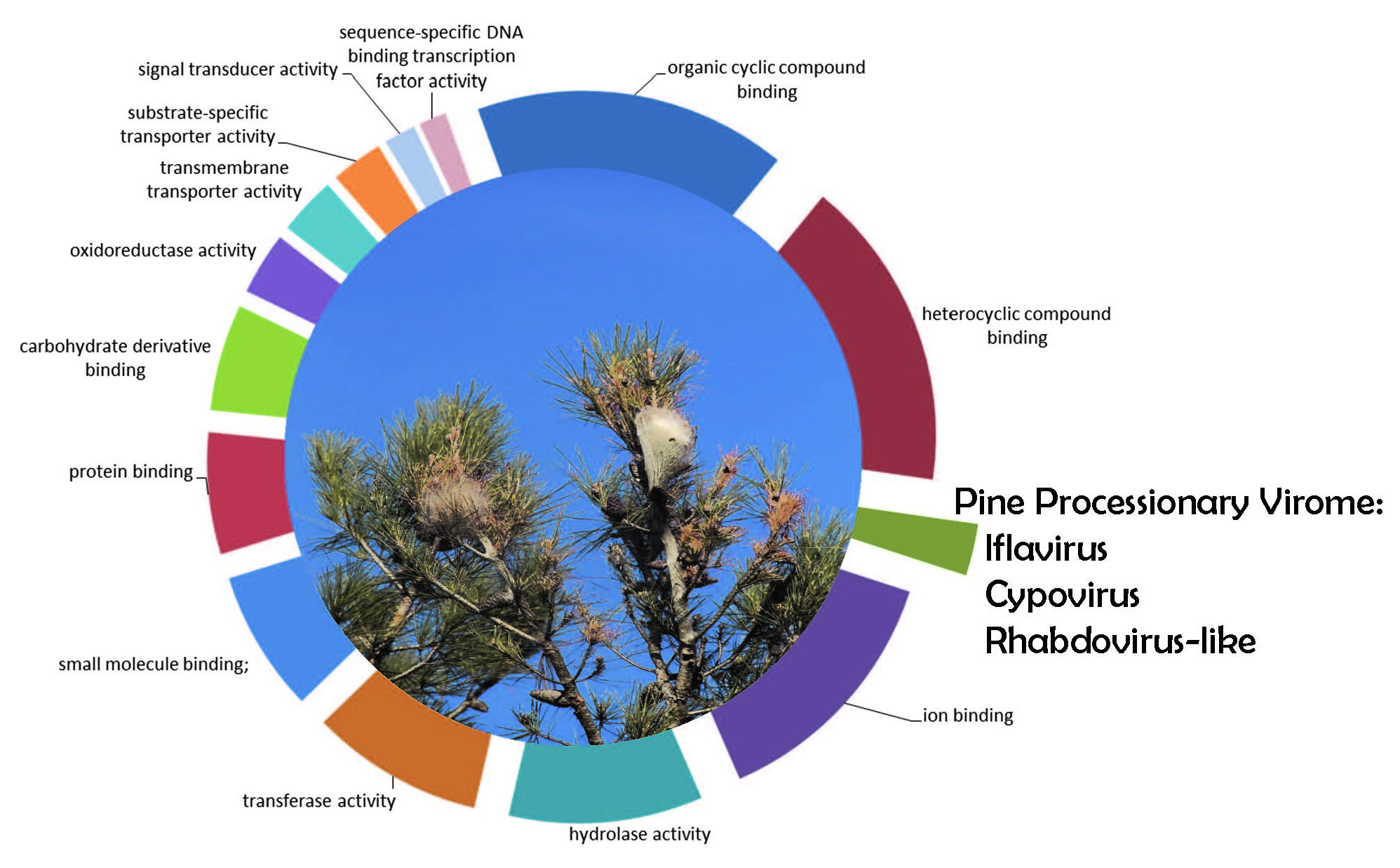{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MG | FB | T | HE | |
|---|---|---|---|---|
| Nr of raw reads | 111,505,976 | 117,988,640 | 114,664,330 | 118,378,516 |
| Total sequence (Mb) | 11,262,103 | 11,916,852 | 11,581,097 | 11,956,230 |
| Sequence quality average | 34 | 34 | 34 | 34 |
| Nr of processed reads | 78,680,476 | 87,668,878 | 86,497,974 | 89,424,098 |
| Sequence quality average a | 36 | 36 | 36 | 36 |
| Nr of unigenes | 152,669 |
| Nr of transcripts | 161,682 |
| Average (median) transcript length | 610 (322) |
| Min-Max transcript length | 201–49,848 |
| N50 transcript length a | 924 |
| Total nr of residues | 98,648,698 |



3.2. Identification of Viral Sequences
| Virus | Unigene | Sequence Length | BLAST Match | e-Value |
|---|---|---|---|---|
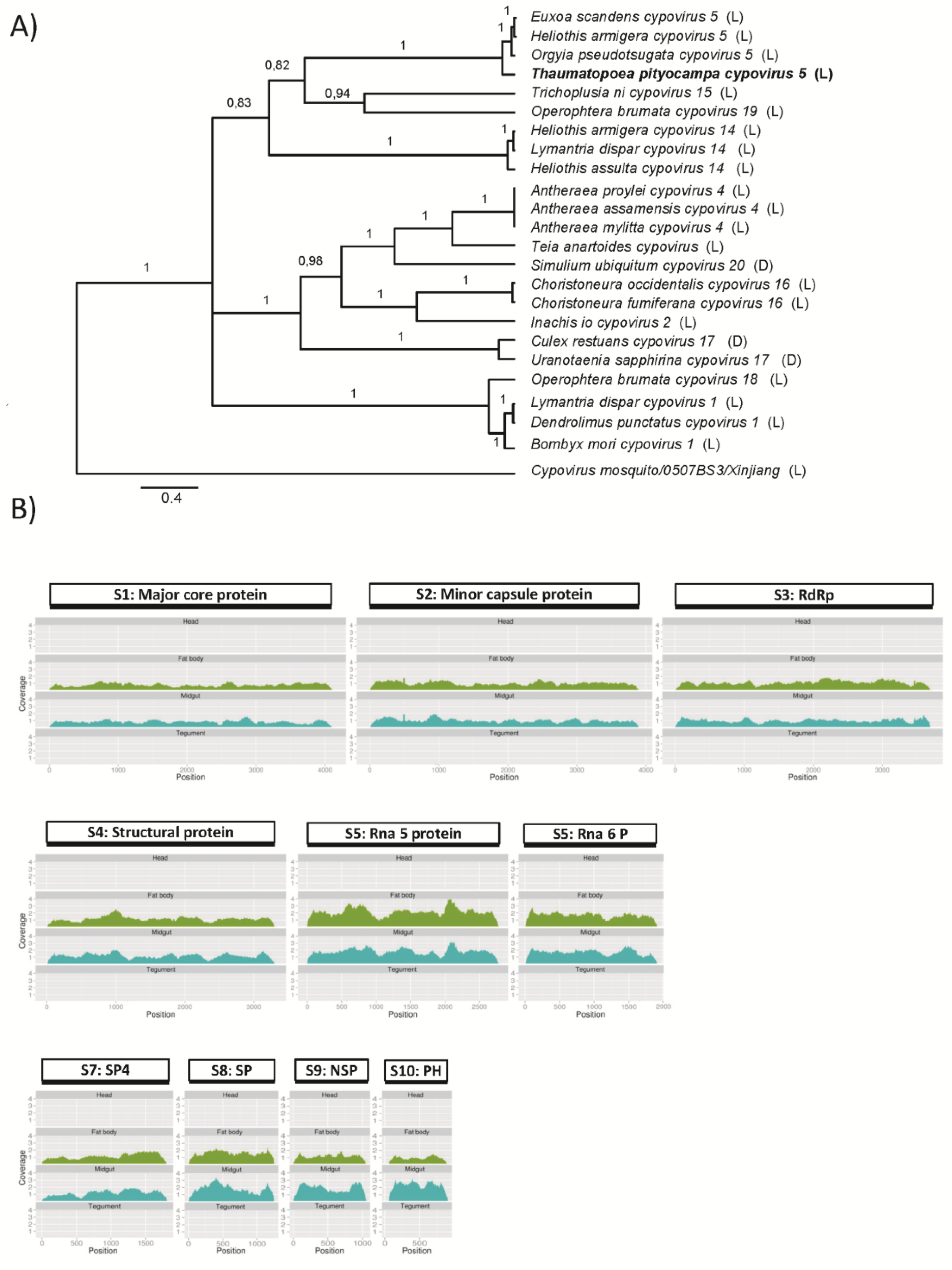
| Cytoplasmic polyhedrosis virus | TPUC35905_TC01 | 4102 | Heliothis armigera CPV5 major core protein | 0.0 |
| TPUC31106_TC01 | 3898 | Orgyia pseudotsugata CPV5 minor capsid protein | 0.0 | |
| TPUC32941_TC01 | 3698 | Orgyia pseudotsugata CPV5 RdRp | 0.0 | |
| TPUC35768_TC01 | 3304 | Orgyia pseudotsugata CPV5 Structural protein | 0.0 | |
| TPUC32710_TC01 | 2780 | Heliothis armigera CPV5 Rna 5 protein | 0.0 | |
| TPUC25109_TC01 | 1813 | Orgyia pseudotsugata CPV5Rna 6 protein | 0.0 | |
| TPUC37981_TC01 | 1914 | Orgyia pseudotsugata CPV5 Viral structural protein 4 (SP4) | 0.0 | |
| TPUC80863_TC01 | 1249 | Heliothis armigera CPV5 Structural protein (SP) | 0.0 | |
| TPUC98042_TC01 | 1058 | Heliothis armigera CPV5 Non-structural protein (NSP) | 0.0 | |
| TPUC71859_TC01 | 924 | Orgyia pseudotsugata CPV5 Polyhedrin (PH) | 1.60E-175 | |
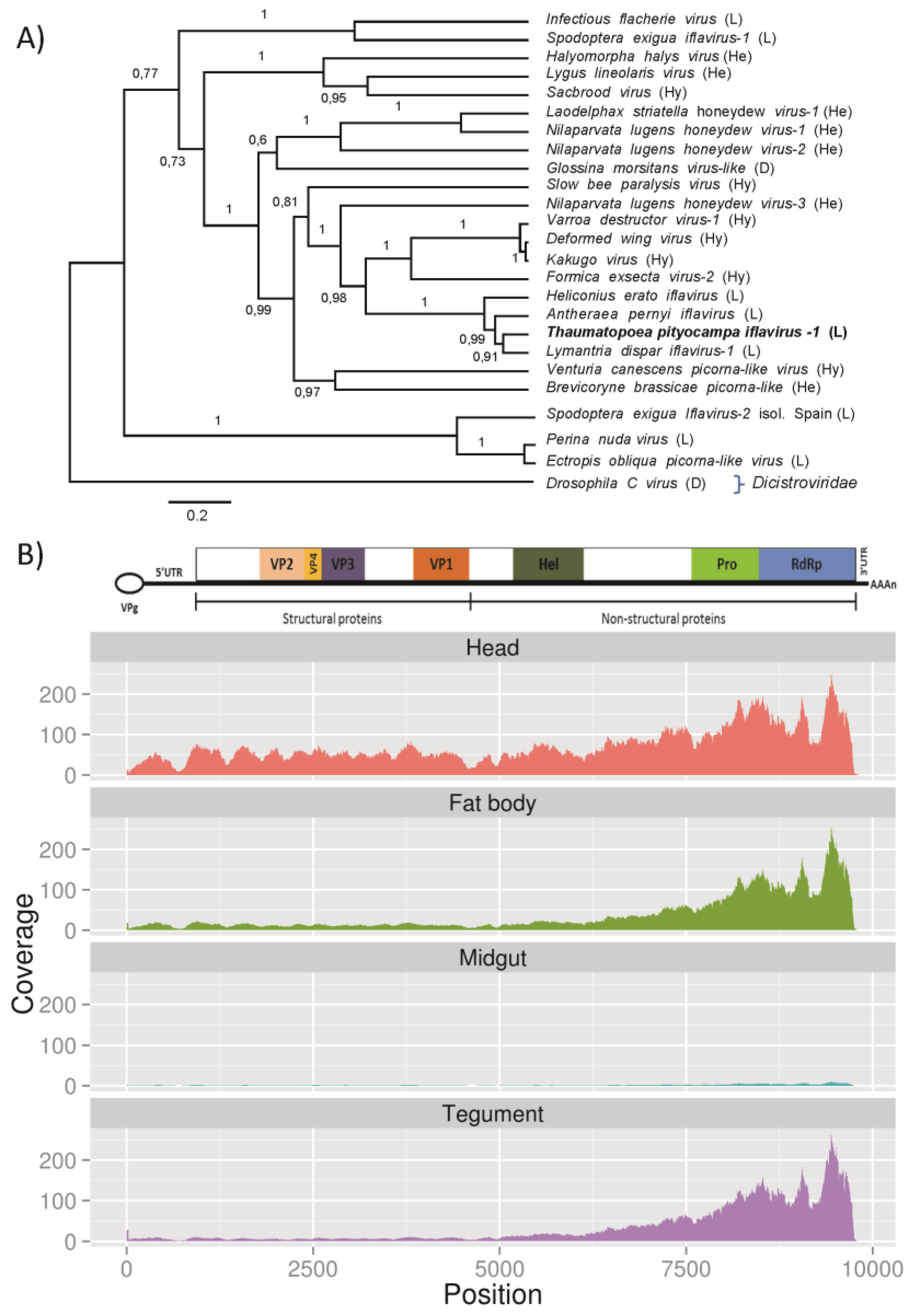
| Iflavirus | TPUC51699_TC01 | 9816 | Antheraea pernyi iflavirus polyprotein | 0.0 |
| TPUC109136_TC01 | 357 | S.exigua iflavirus 2 polyprotein | 3.30E-58 | |
| TPUC143735_TC01 | 335 | Antheraea pernyi iflavirus polyprotein | 2.35E-18 | |
| TPUC71578_TC01 | 328 | S.exigua iflavirus 2 polyprotein | 1.26E-49 | |
| TPUC147958_TC01 | 323 | Antheraea pernyi iflavirus polyprotein | 1.15E-07 | |
| TPUC03472_TC01 | 298 | S.exigua iflavirus 2 polyprotein | 9.18E-56 | |
| TPUC100622_TC01 | 230 | S.exigua iflavirus 2 polyprotein | 3.18E-39 | |
| TPUC83511_TC01 | 225 | S.exigua iflavirus 2 polyprotein | 1.82E-37 | |
| TPUC151007_TC01 | 217 | S.exigua iflavirus 2 polyprotein | 3.84E-23 | |
| TPUC91335_TC01 | 214 | S.exigua iflavirus 2 polyprotein | 1.36E-37 | |
| Rhabdovirus | TPUC44929_TC01 | 4501 | Maraba virus L polymerase protein | 0.0 |
| TPUC38841_TC01 | 2830 | Spodoptera frugiperda rhabdovirus P protein | 6.50E-11 | |
| TPUC14459_TC01 | 1900 | Jurona virus L polymerase protein | 1.09E-115 | |
| TPUC75494_TC01 | 1016 | Dolphin rhabdovirus L polymerase protein | 7.40E-12 | |
| TPUC37175_TC01 | 914 | Muscina stabulans sigmavirus RdRp | 5.31E-48 | |
| TPUC48042_TC01 | 895 | Drosophila immigrans sigmavirus RdRp | 1.35E-15 | |
| TPUC98122_TC01 | 631 | Muscina stabulans sigmavirus RdRp | 1.51E-55 | |
| TPUC56532_TC01 | 516 | China fish rhabdovirus L polymerase protein | 7.95E-72 | |
| TPUC99390_TC01 | 361 | Muscina stabulans sigmavirus RdRp | 2.79E-34 |
3.2.1. T. pityocampa Cytoplasmic Polyhedrosis Virus, TpCPV 5

3.2.2. T. pityocampa Iflavirus, TpIV1
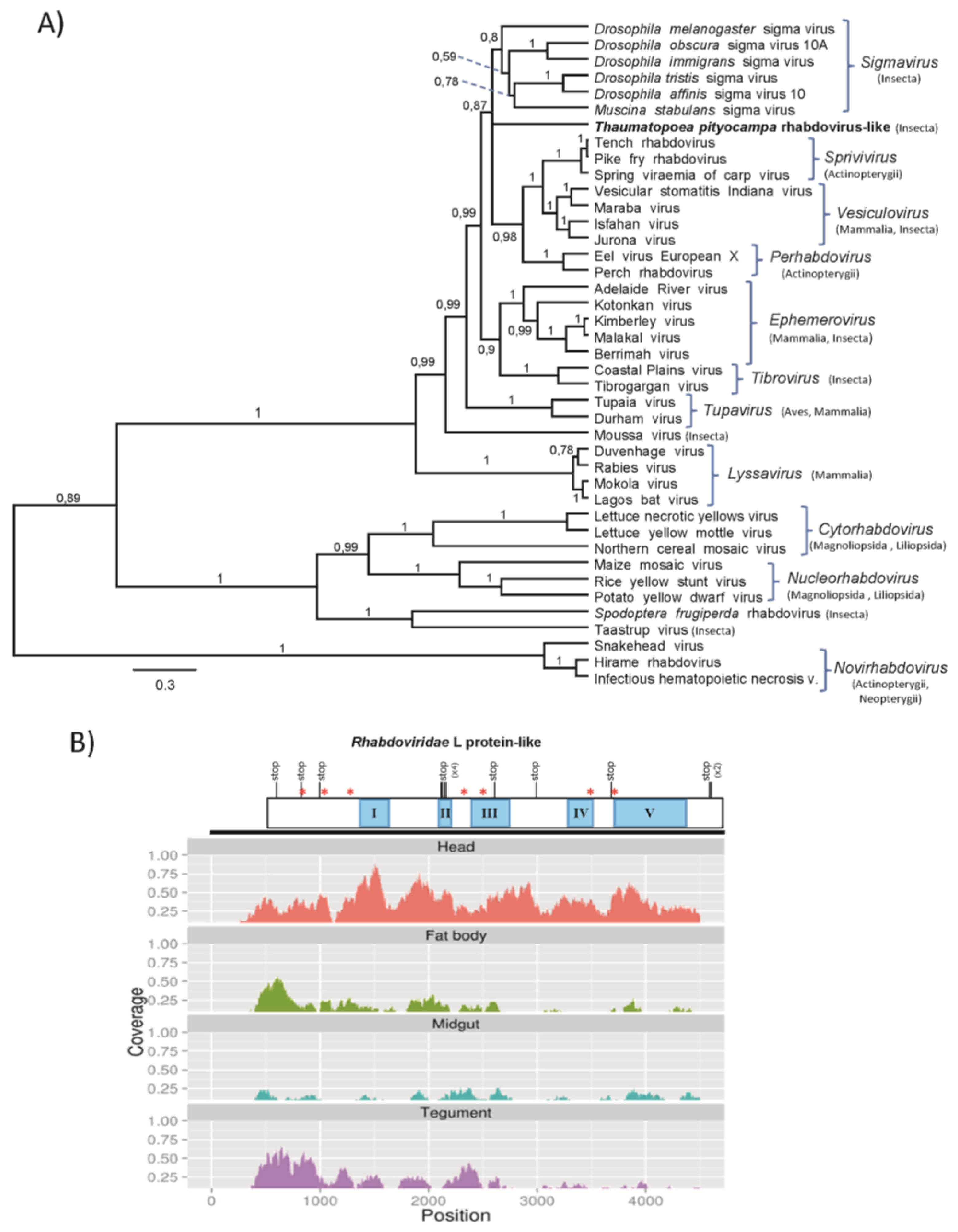
3.2.3. T. pityocampa Rhabdovirus-Like Sequences


4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vega, J.M.; Moneo, I.; Armentia, A.; Lopez-Rico, R.; Curiel, G.; Bartolome, B.; Fernandez, A. Anaphylaxis to a pine caterpillar. Allergy: Eur. J. Allergy Clin. Immunol. 1997, 52, 1244–1245. [Google Scholar] [CrossRef]
- Rousselet, J.; Imbert, C.E.; Dekri, A.; Garcia, J.; Goussard, F.; Vincent, B.; Denux, O.; Robinet, C.; Dorkeld, F.; Roques, A.; et al. Assessing species distribution using Google Street View: A pilot study with the pine processionary moth. PLoS One 2013, 8, e74918. [Google Scholar] [CrossRef] [PubMed]
- Forestry Commission. Available online: http://www.forestry.gov.uk/pineprocessionarymoth (accessed on 20 November 2014).
- Atakan, A. Biology of harmful insects of Forests in Turkey; Forest General Directorate Publication: Ankara, Tyrkey, 1991. [Google Scholar]
- Masutti, L.; Battisti, A. Thaumetopoea pityocampa (Den. & Schiff.) in Italy Bionomics and perspectives of integrated control12. J. Appl. Entomol. 1990, 110, 229–234. [Google Scholar] [CrossRef]
- Kumar, S.; Chandra, A.; Pandey, K.C. Bacillus thuringiensis (Bt) transgenic crop: An environment friendly insect-pest management strategy. J. Environ. Biol. 2008, 29, 641–653. [Google Scholar] [PubMed]
- Cebeci, H.H.; Oymen, R.T.; Acer, S. Control of pine processionary moth, Thaumetopoea pityocampa with Bacillus thuringiensis in Antalya, Turkey. J. Environ. Biol 2010, 31, 357–361. [Google Scholar] [PubMed]
- Navon, A. Bacillus thuringiensis insecticides in crop protection—Reality and prospects. Crop Prot. 2000, 19, 669–676. [Google Scholar] [CrossRef]
- Shevelev, A.B.; Battisti, A.; Volynskaya, A.M.; Inovikova, S.; Kostina, L.I.; Zalunin, I.A. Susceptibility of the pine processionary caterpillar Thaumetopoea pityocampa (Lepidoptera: Thaumetopoeidae) toward d-endotoxins of Bacillus thuringiensis under laboratory conditions. Ann. Appl. Biol. 2001, 138, 255–261. [Google Scholar] [CrossRef]
- Gatto, P.; Zocca, A.; Battisti, A.; Barrento, M.J.; Branco, M.; Paiva, M.R. Economic assessment of managing processionary moth in pine forests: A case-study in Portugal. J. Environ. Manag. 2009, 90, 683–691. [Google Scholar] [CrossRef]
- Ince, I.A.; Demir, I.; Demirbag, Z.; Nalcacioglu, R. A cytoplasmic polyhedrosis virus isolated from the pine processionary caterpillar, Thaumetopoea pityocampa. J. Microbiol. Biotechnol. 2007, 17, 632–637. [Google Scholar] [PubMed]
- Sevim, A.; Demir, I.; Demirbag, Z. Molecular characterization and cirulence of Beauveria spp. from the pine processionary moth, Thaumetopoea pityocampa (Lepidoptera: Thaumetopoeidae). Mycopathologia 2010, 170, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Rastall, K.; Kondo, V.; Strazanac, J.S.; Butler, L. Lethal effects of biological insecticide applications on nontarget lepidopterans in two appalachian forests. Environ. Entomol. 2003, 32, 1364–1369. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. Metagenomic analysis of coastal RNA virus communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Studholme, D.J.; Glover, R.H.; Boonham, N. Application of high-throughput DNA sequencing in phytopathology. Ann. Rev. Phytopathol. 2011, 49, 87–105. [Google Scholar] [CrossRef]
- Liu, S.; Vijayendran, D.; Bonning, B.C. Next generation sequencing technologies for insect virus discovery. Viruses 2011, 3, 1849–1869. [Google Scholar] [CrossRef] [PubMed]
- Zirkel, F.; Kurth, A.; Quan, P.L.; Briese, T.; Ellerbrok, H.; Pauli, G.; Leendertz, F.H.; Lipkin, W.I.; Ziebuhr, J.; Drosten, C.; et al. An insect nidovirus emerging from a primary tropical rainforest. MBIO 2011, 2, e00077-11. [Google Scholar] [CrossRef] [PubMed]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.L.; Briese, T.; Hornig, M.; Geiser, D.M.; et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef] [PubMed]
- vanengelsdorp, D.; Meixner, M.D. A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 2010, 103, S80–S95. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Gao, F.; Ye, X.Q.; Wei, S.J.; Shi, M.; Zheng, H.J.; Chen, X.X. Deep sequencing of Cotesia vestalis bracovirus reveals the complexity of a polydnavirus genome. Virology 2011, 414, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Abd-Alla, A.M.M.; Cousserans, F.; Parker, A.G.; Jehle, J.A.; Parker, N.J.; Vlak, J.M.; Robinson, A.S.; Bergoin, M. Genome analysis of a Glossina pallidipes salivary gland hypertrophy virus reveals a novel, large, double-stranded circular DNA virus. J. Virol. 2008, 82, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Pascual, L.; Jakubowska, A.K.; Blanca, J.M.; Cañizares, J.; Ferré, J.; Gloeckner, G.; Vogel, H.; Herrero, S. The transcriptome of Spodoptera exigua larvae exposed to different types of microbes. Insect Biochem. Mol. Biol. 2012, 42, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Gschloessl, B.; Vogel, H.; Burban, C.; Heckel, D.; Streiff, R.; Kerdelhue, C. Comparative analysis of two phenologically divergent populations of the pine processionary moth (Thaumetopoea pityocampa) by de novo transcriptome sequencing. Insect Biochem. Mol. Biol. 2014, 46, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Winnebeck, E.C.; Millar, C.D.; Warman, G.R. Why does insect RNA look degraded? J. Insect Sci. 2010, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics. FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 November 2014).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature 2011, 29, 644–652. [Google Scholar]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A Program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol 1999, 138–148. [Google Scholar]
- Li, Y.; Tan, L.; Li, Y.; Chen, W.; Zhang, J.; Hu, Y. Identification and genome characterization of Heliothis armigera cypovirus types 5 and 14 and Heliothis assulta cypovirus type 14. J. Gen. Virol. 2006, 87, 387–394. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Adams, M.J.; Lefkowitz, E.J.; Carstens, E.B. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Shapiro, A.; Green, T.; Rao, S.; White, S.; Carner, G.; Mertens, P.P.; Becnel, J.J. Morphological and molecular characterization of a cypovirus (Reoviridae) from the mosquito Uranotaenia sapphirina (Diptera: Culicidae). J. Virol. 2005, 79, 9430–9438. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Simossis, V.A.; Heringa, J. PRALINE: A multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005, 33 (Suppl. 2), W289–W294. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-Based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Bourhy, H.; Cowley, J.A.; Larrous, F.; Holmes, E.C.; Walker, P.J. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 2005, 86, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, K.B.; Nicholas, H.B. J.; Deerfield, D.W. GeneDoc: Analysis and visualization of genetic variation. EMBNEW. NEWS 1997, 4, 14. [Google Scholar]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- BEAST Software—Bayesian Evolutionary Analysis Sampling Trees. Available online: http://beast.bio.ed.ac.uk/programs (accessed on 20 November 2014).
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.O. Survey of new data and computer methods of analysis. In Atlas of Protein Sequence and Structure; Dayhoff, M.O., Ed.; National Biomedical Research Foundation: Washington, DC, USA, 1976. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2009. [Google Scholar]
- Finseth, F.R.; Harrison, R.G. A comparison of next generation sequencing technologies for transcriptome assembly and utility for RNA-Seq in a non-model bird. PLoS One 2014, 9, e108550. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahrour, F.; Diaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Mertens, P.P.C.; Estes, M.K.; Arella, M.; Attoui, H.; Belloncik, S.; Bergoin, M.; Boccardo, G.; Booth, T.F.; Chiu, W.; Diprose, J.M.; et al. Virus Taxonomy: Classification and Nomenclature of Viruses: Seventh Report of the International Committee on Taxonomy of Viruses; Academic Press: San Diego, CA, USA, 2000; pp. 395–480. [Google Scholar]
- Hukuhara, T. Reoviridae. In Atlas of Invertebrate Viruses; Adams, J.R., Bonami, J.R., Eds.; CRC Press: Boca Raton, FL, USA, 1991; pp. 393–434. [Google Scholar]
- Eberle, K.E.; Wennman, J.T.; Klespies, R.G.; Jehle, J.A. Basic techniques in insect virology. In Manual. of Techniques. in Invertebrate Pathology, 2nd ed.; Academic: London, UK, 2012; pp. 15–74. [Google Scholar]
- Hu, Z.; Chen, X.; Sun, X. Molecular biology of insect viruses. In Advances in Microbial Control of Insect Pests; Springer: Berlin, Germany, 2003; pp. 83–107. [Google Scholar]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/virusTaxonomy.asp?version=2009 (accessed on 20 November 2014).
- Van Oers, M.M. Iflavirus. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., van Regenmortel, M.H.V., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2008; Volume 3, pp. 42–46. [Google Scholar]
- Geng, P.; Li, W.; Lin, L.; de Miranda, J.R.; Emrich, S.; An, L.; Terenius, O. Genetic characterization of a novel iflavirus associated with vomiting disease in the chinese oak silkmoth Antheraea pernyi. PLoS One 2014, 9, e92107. [Google Scholar] [CrossRef] [PubMed]
- Ongus, J.R.; Roode, E.C.; Pleij, C.W.; Vlak, J.M.; van Oers, M.M. The 5′ non-translated region of Varroa destructor virus 1 (genus Iflavirus): Structure prediction and IRES activity in Lymantria dispar cells. J. Gen. Virol. 2006, 87, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Fievet, J.; Tentcheva, D.; Gauthier, L.; de, M.J.; Cousserans, F.; Colin, M.E.; Bergoin, M. Localization of deformed wing virus infection in queen and drone Apis mellifera L. Virol. J. 2006, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Jakubowska, A.K.; D’Angiolo, M.; Gonzáez-Martínez, R.M.; Millán-Leiva, A.; Carballo, A.; Murillo, R.; Caballero, P.; Herrero, S. Simultaneous occurrence of covert infections with small RNA viruses in the lepidopteran Spodoptera exigua. J. Invertebr. Pathol. 2014, 121, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Tripp, J.; Krueger, E.N.; Harrison, R.L.; Toth, A.L.; Allen Miller, W.; Bonning, B.C. Lymantria dispar iflavirus 1 (LdIV1), a new model to study iflaviral persistence in lepidopterans. J. Gen. Virol. 2014, 95, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Macias-Muñoz, A.; Briscoe, A.D. Genome sequence of a novel Iflavirus from mRNA sequencing of the butterfly Heliconius erato. Genome Announc. 2014, 2, e00398-14. [Google Scholar] [PubMed]
- Zioni, N.; Soroker, V.; Chejanovsky, N. Replication of Varroa destructor virus 1 (VDV-1) and a Varroa destructor virus 1-deformed wing virus recombinant (VDV-1-DWV) in the head of the honey bee. Virology 2011, 417, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Terio, V.; Martella, V.; Camero, M.; Decaro, N.; Testini, G.; Bonerba, E.; Tantillo, G.; Buonavoglia, C. Detection of a honeybee iflavirus with intermediate characteristics between kakugo virus and deformed wing virus. New Microbiol. 2008, 31, 439–444. [Google Scholar] [PubMed]
- Aizawa, K.; Kuruta, Y. Infection under aseptic conditions with the virus of infectious flacherie in the silkworm, Bombyx mori. J. Insect Pathol. 1964, 6, 130–132. [Google Scholar]
- Choi, H.K.; Kobayashi, M.; Kawase, S. Changes in infectious flacherie virus-specific polypeptides and translatable mRNA in the midgut of the silkworm, Bombyx mori, during larval molt. J. Invertebr. Pathol. 1989, 53, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Fujiyuki, T.; Takeuchi, H.; Ono, M.; Ohka, S.; Sasaki, T.; Nomoto, A.; Kubo, T. Kakugo Virus from Brains of Aggressive Worker Honeybees. Adv. Virus Res. 2005, 65, 1–27. [Google Scholar] [PubMed]
- Wagner, R.R.; Prevec, L.; Brown, F.; Summers, D.F.; Sokol, F.; MacLeod, R. Classification of Rhabdovirus proteins: A proposal. J. Virol. 1972, 10, 1228–1230. [Google Scholar]
- Poch, O.; Blumberg, B.M.; Bougueleret, L.; Tordo, N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: Theoretical assignment of functional domains. J. Gen. Virol. 1990, 71, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, M.J.; Bruenn, J.A.; Taylor, D.J. Phylogeny, integration and expression of sigma virus-like genes in Drosophila. Mol. Phylogen. Evol. 2012, 65, 251–258. [Google Scholar] [CrossRef]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PloS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- Fort, P.; Albertini, A.; van-Hua, A.; Berthomieu, A.; Roche, S.; Delsuc, F.; Pasteur, N.; Capy, P.; Gaudin, Y.; Weill, M. Fossil rhabdoviral sequences integrated into arthropod genomes: Ontogeny, evolution, and potential functionality. Mol. Biol. Evol. 2012, 29, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.J.; Dittmar, K.; Ballinger, M.J.; Bruenn, J.A. Evolutionary maintenance of filovirus-like genes in bat genomes. BMC Evol. Biol. 2011, 11, e336. [Google Scholar] [CrossRef]
- Taylor, D.J.; Bruenn, J. The evolution of novel fungal genes from non-retroviral RNA viruses. BMC Biol. 2009, 7, e88. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Novella, I.S.; Dietzgen, R.G.; Padhi, A.; Rupprecht, C.E. The rhabdoviruses: Biodiversity, phylogenetics, and evolution. Infect. Genet. Evol. 2009, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Hogenhout, S.A.; Redinbaugh, M.G.; Ammar, E.D. Plant and animal rhabdovirus host range: A bug’s view. Trends Microbiol. 2003, 11, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.A.; Obbard, D.J.; Maside, X.; Jiggins, F.M. The recent spread of a vertically transmitted virus through populations of Drosophila melanogaster. Mol. Ecol. 2007, 16, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Longdon, B.; Hadfield, J.D.; Webster, C.L.; Obbard, D.J.; Jiggins, F.M. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathog. 2011, 7, e1002260. [Google Scholar] [CrossRef]
- Longdon, B.; Obbard, D.J.; Jiggins, F.M. Sigma viruses from three species of Drosophila form a major new clade in the rhabdovirus phylogeny. Proc. R. Soc. Lond. B Biol. Sci. 2010, 277, 35–44. [Google Scholar] [CrossRef]
- Ma, H.; Galvin, T.A.; Glasner, D.R.; Shaheduzzaman, S.; Khan, A.S. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J. Virol. 2014, 88, 6576–6585. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.O.; Lundsgaard, T.; Pedersen, P.A.; Christensen, L.S. Identification and partial characterization of Taastrup virus: A newly identified member species of the Mononegavirales. Virology 2004, 319, 49–59. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubowska, A.K.; Nalcacioglu, R.; Millán-Leiva, A.; Sanz-Carbonell, A.; Muratoglu, H.; Herrero, S.; Demirbag, Z. In Search of Pathogens: Transcriptome-Based Identification of Viral Sequences from the Pine Processionary Moth (Thaumetopoea pityocampa). Viruses 2015, 7, 456-479. https://doi.org/10.3390/v7020456
Jakubowska AK, Nalcacioglu R, Millán-Leiva A, Sanz-Carbonell A, Muratoglu H, Herrero S, Demirbag Z. In Search of Pathogens: Transcriptome-Based Identification of Viral Sequences from the Pine Processionary Moth (Thaumetopoea pityocampa). Viruses. 2015; 7(2):456-479. https://doi.org/10.3390/v7020456
Chicago/Turabian StyleJakubowska, Agata K., Remziye Nalcacioglu, Anabel Millán-Leiva, Alejandro Sanz-Carbonell, Hacer Muratoglu, Salvador Herrero, and Zihni Demirbag. 2015. "In Search of Pathogens: Transcriptome-Based Identification of Viral Sequences from the Pine Processionary Moth (Thaumetopoea pityocampa)" Viruses 7, no. 2: 456-479. https://doi.org/10.3390/v7020456
APA StyleJakubowska, A. K., Nalcacioglu, R., Millán-Leiva, A., Sanz-Carbonell, A., Muratoglu, H., Herrero, S., & Demirbag, Z. (2015). In Search of Pathogens: Transcriptome-Based Identification of Viral Sequences from the Pine Processionary Moth (Thaumetopoea pityocampa). Viruses, 7(2), 456-479. https://doi.org/10.3390/v7020456