
Lentiviral Vector Mediated Claudin1 Silencing Inhibits Epithelial to Mesenchymal Transition in Breast Cancer Cells


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.3. Cell Colony Formation Assay
2.4. MTT Assay
2.5. Cell Migration Assay
2.6. Cell Invasion Assay
2.7. Western Blot
2.8. TCGA Database Query
2.9. Statistical Analysis
3. Results
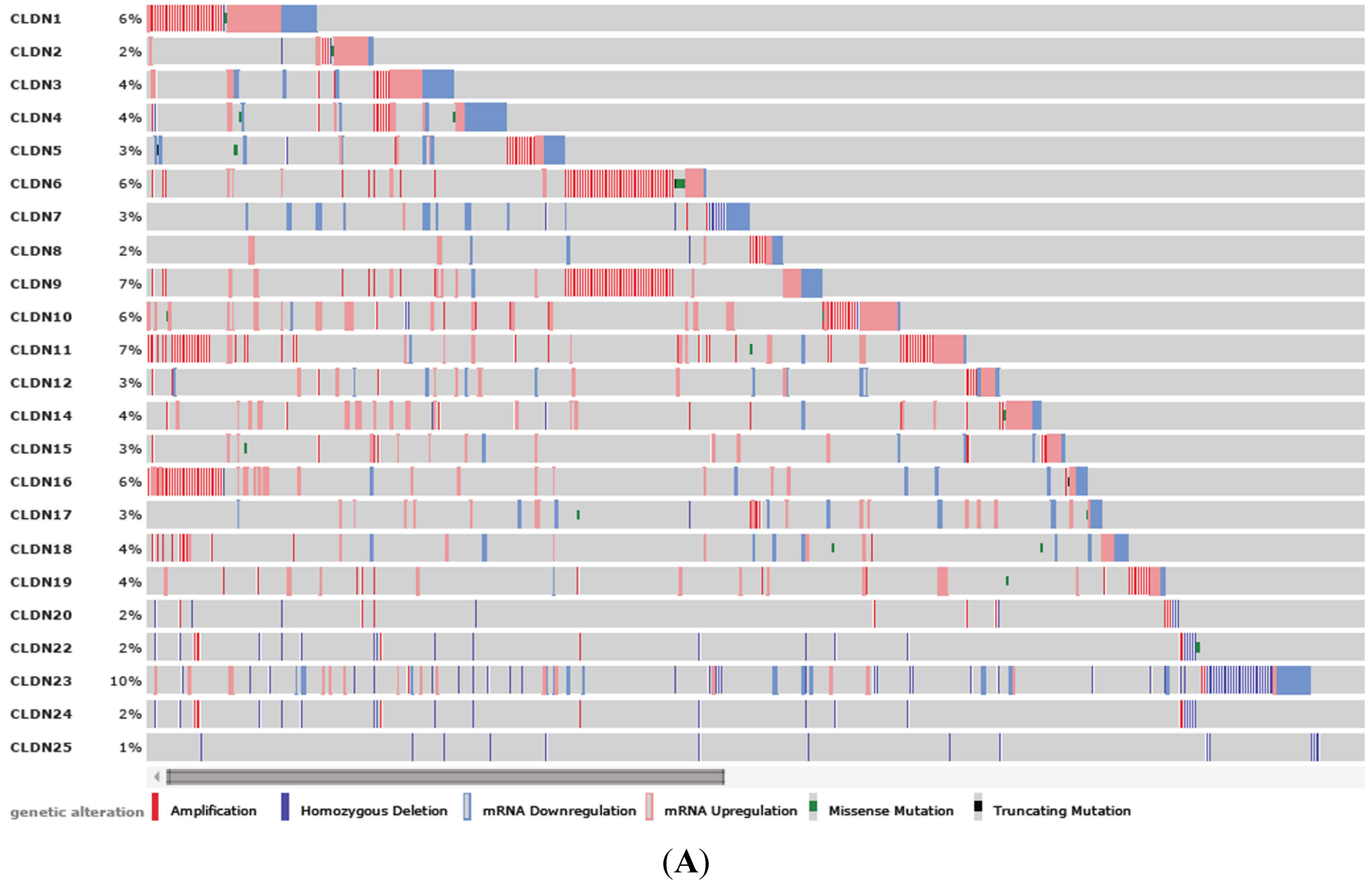
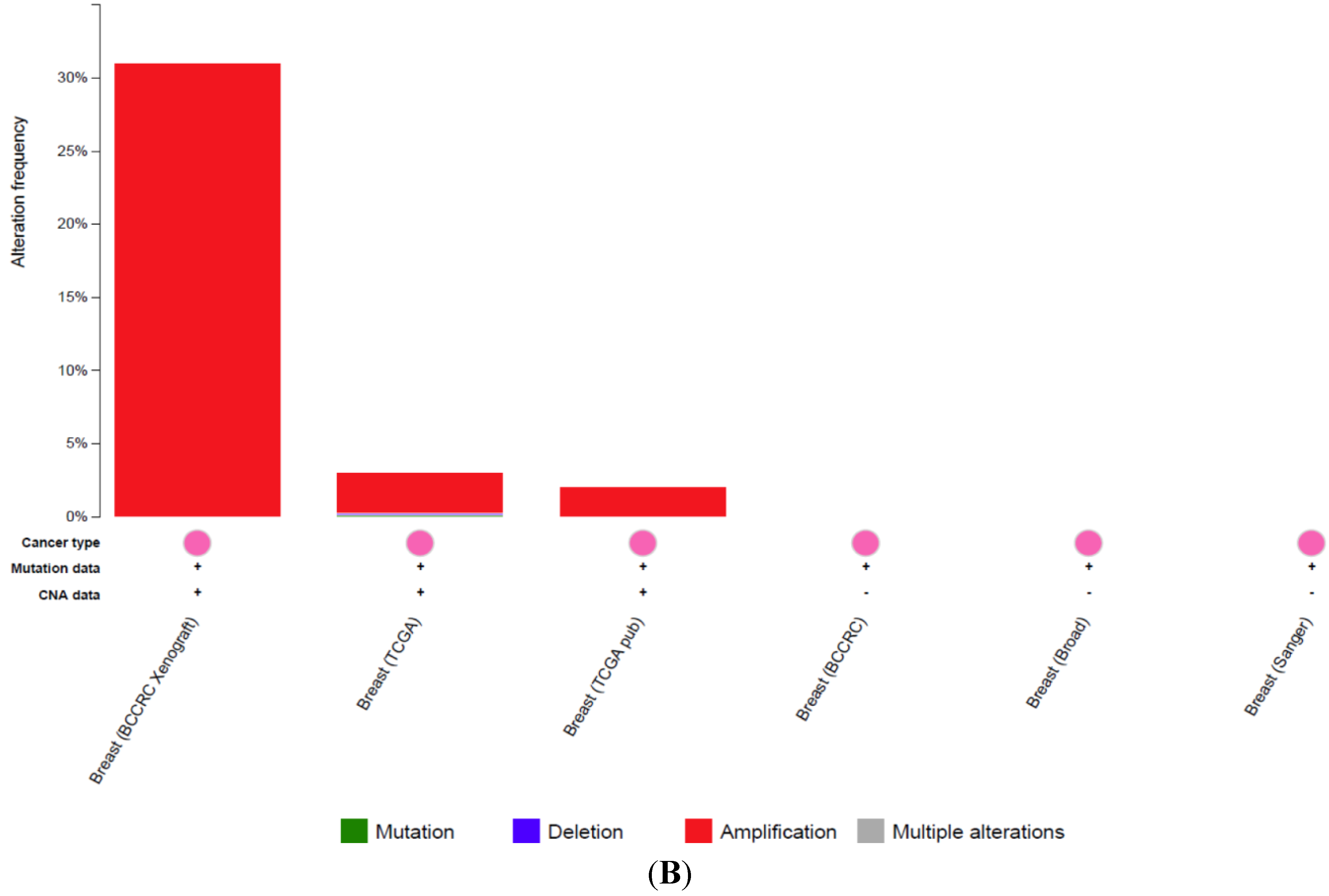
3.1. Clinical Significance of CLDN1 Expression in Breast Cancer


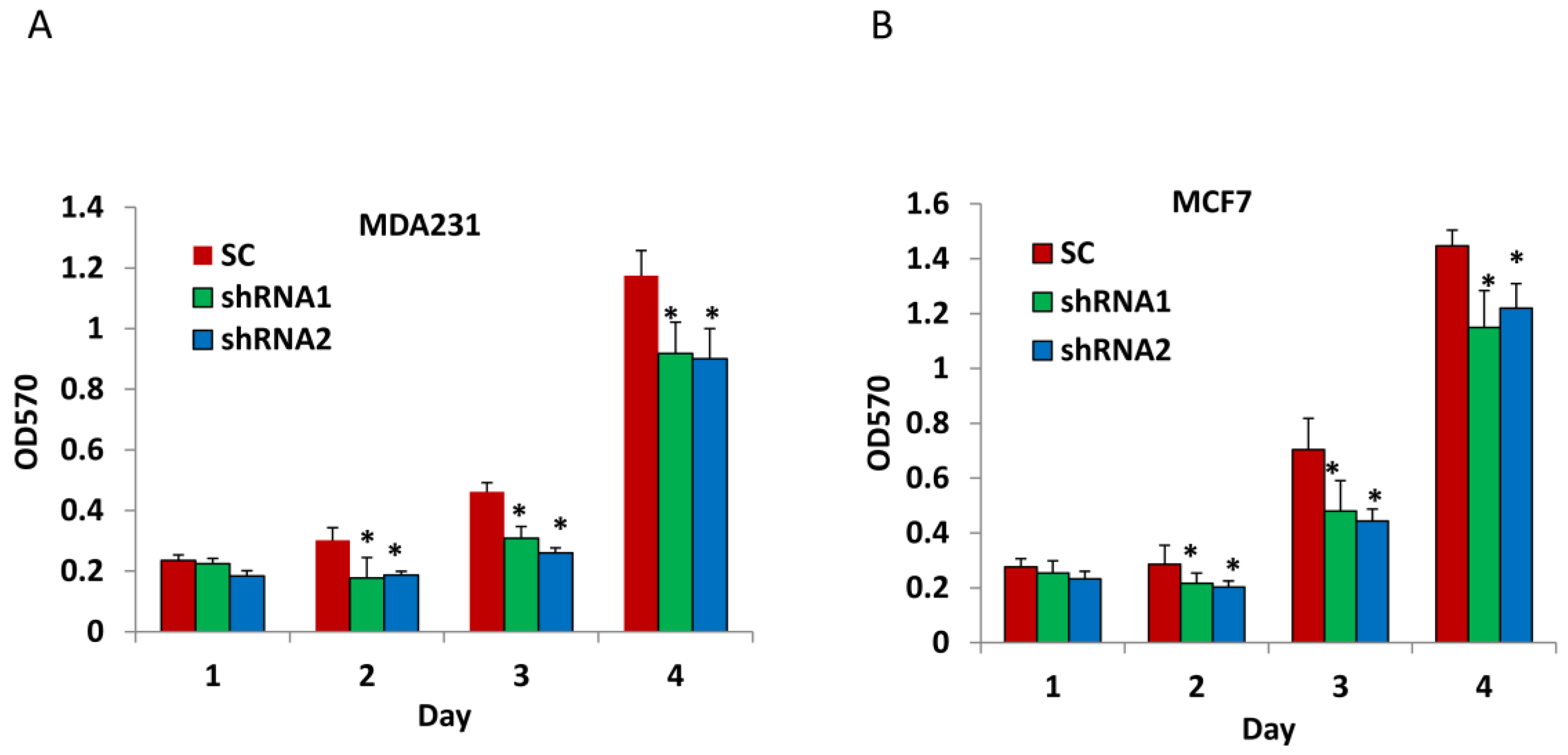
3.2. Silencing CLDN1 Inhibits Breast Cancer Cell Proliferation
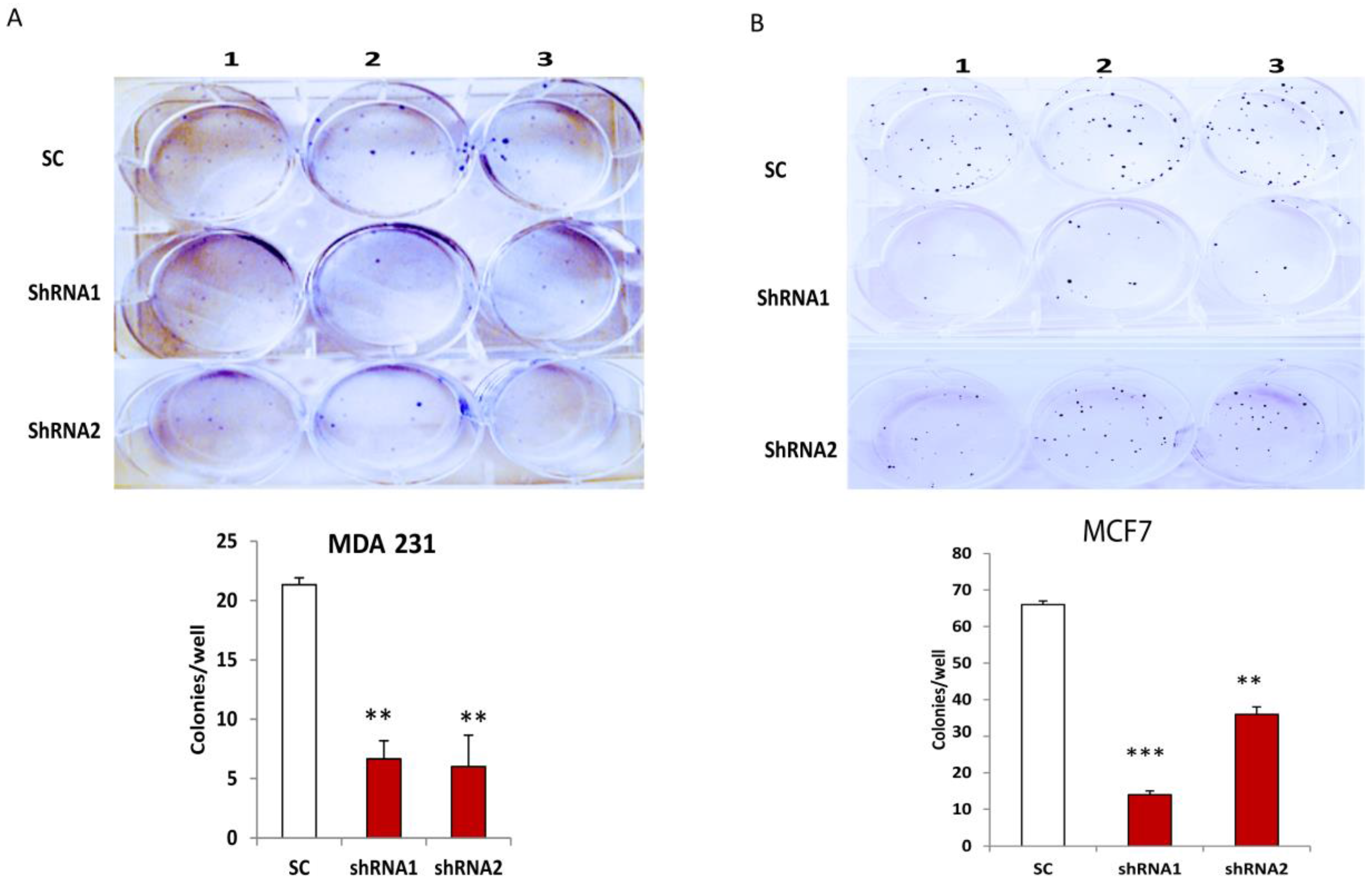
3.3. Silencing CLDN1 Inhibits Clonogenicity of Breast Cancer Cells


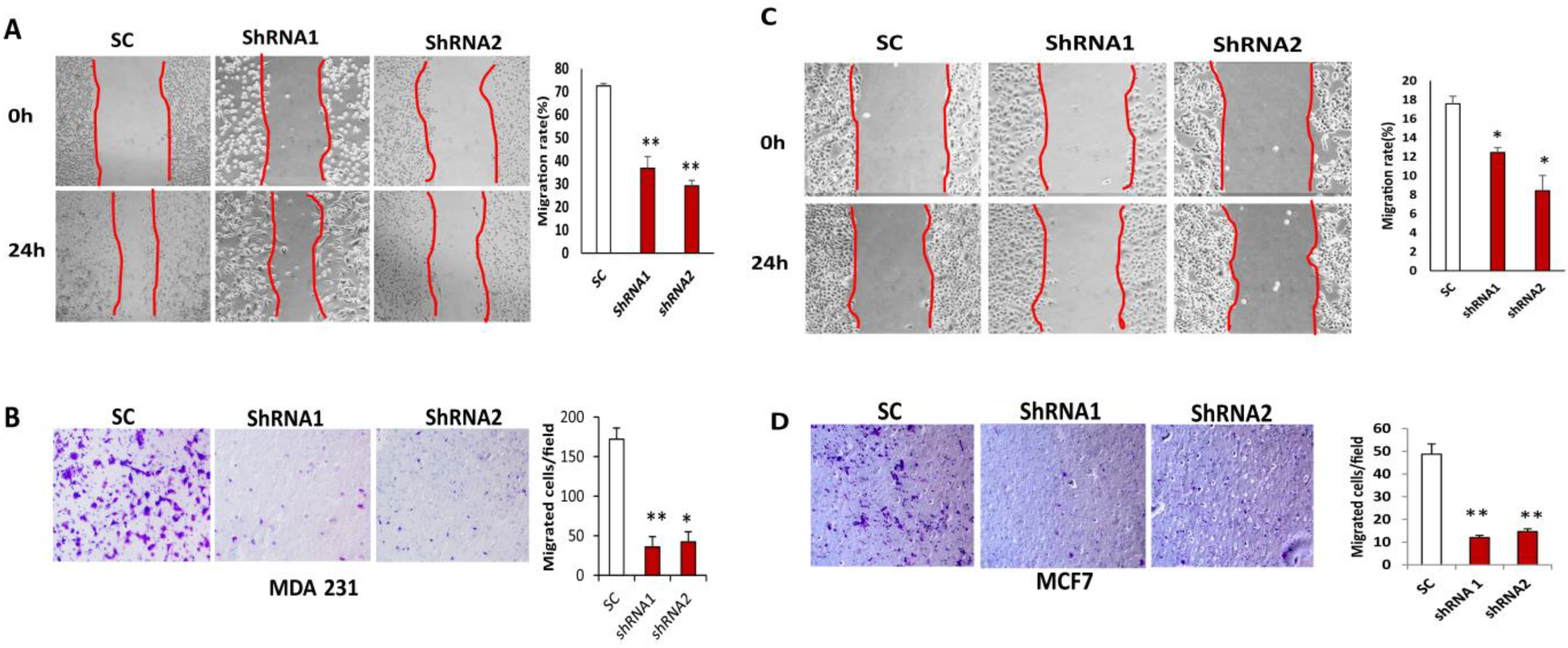
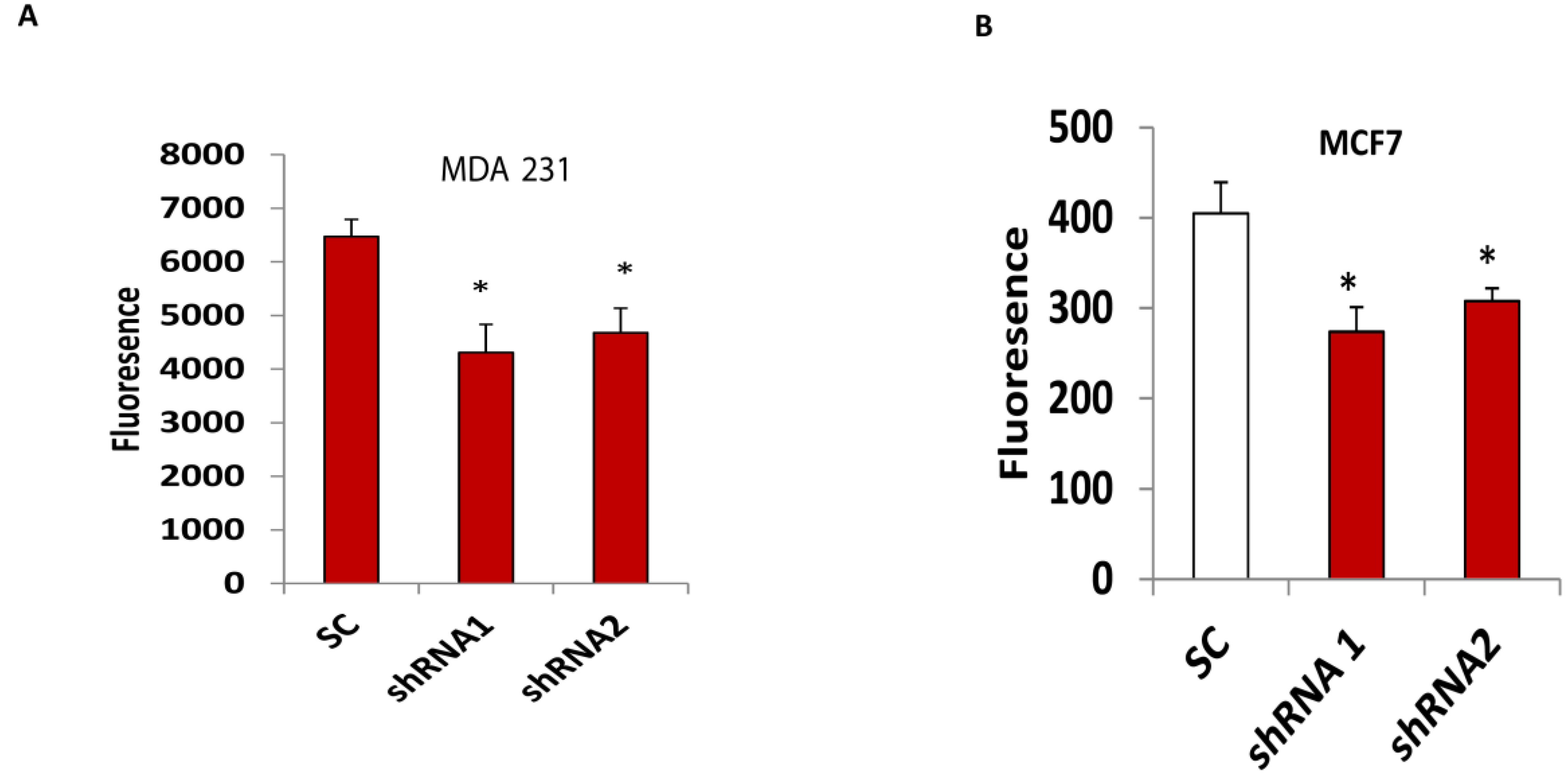
3.4. Silencing CLDN1 Inhibits Breast Cancer Cell Migration and Invasion


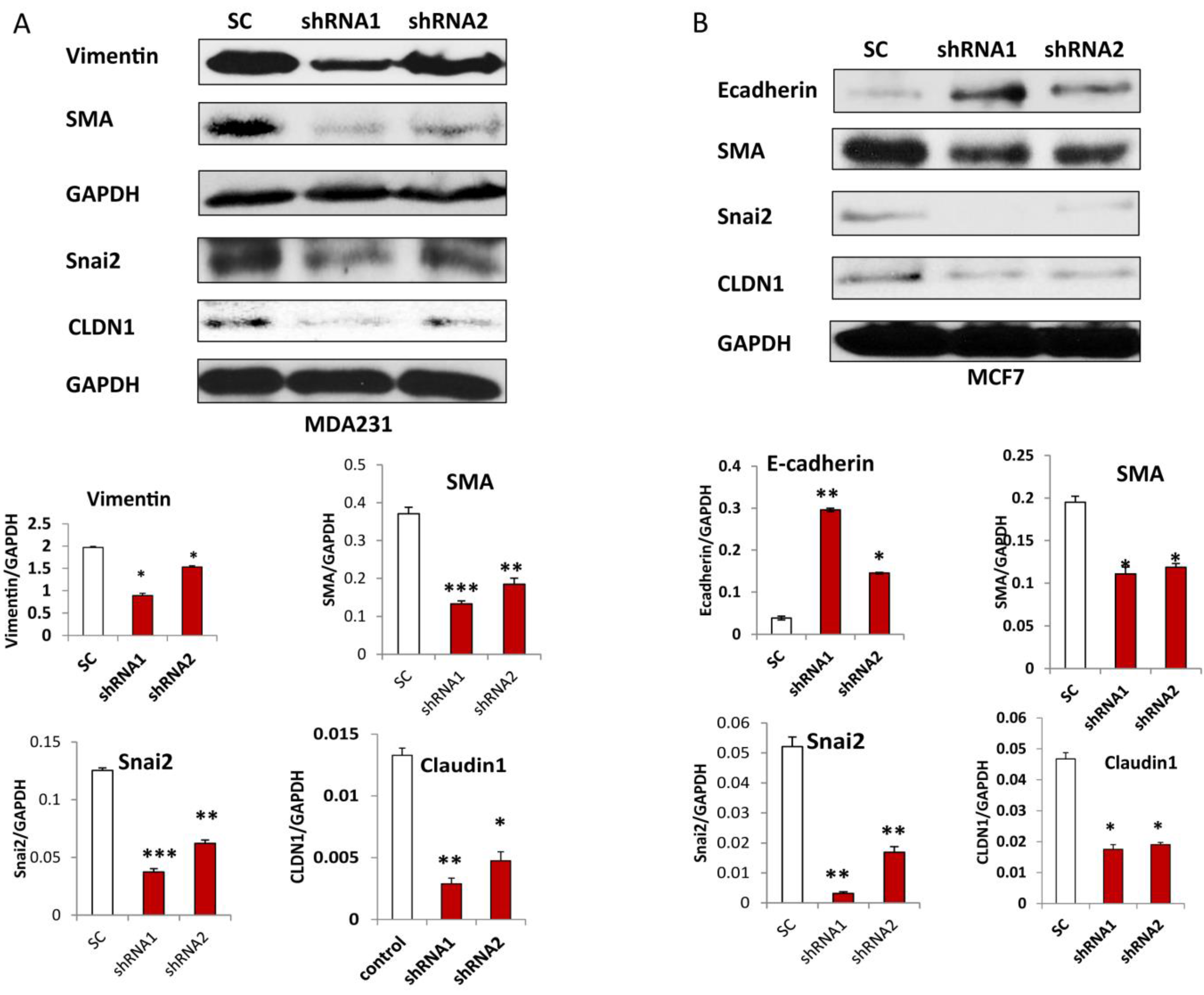
3.5. Silencing CLDN1 Inhibits EMT in Breast Cancer Cells
4. Discussion

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chatenoud, L.; Bertuccio, P.; Bosetti, C.; Malvezzi, M.; Levi, F.; Negri, E.; la Vecchia, C. Trends in mortality from major cancers in the Americas: 1980–2010. Ann. Oncol. 2014, 25, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hicks, M.J.; Kaminsky, S.M.; Moore, M.A.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Majhen, D.; Calderon, H.; Chandra, N.; Fajardo, C.A.; Rajan, A.; Alemany, R.; Custers, J. Adenovirus-based vaccines for fighting infectious diseases and cancer: Progress in the field. Hum. Gene Ther. 2014, 25, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.K.; Chiocca, E.A. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014, 16, 334–351. [Google Scholar] [CrossRef] [PubMed]
- Zolotukhin, I.; Luo, D.; Gorbatyuk, O.; Hoffman, B.; Warrington, K., Jr.; Herzog, R.; Harrison, J.; Cao, O. Improved Adeno-associated Viral Gene Transfer to Murine Glioma. J. Genet. Syndr. Gene Ther. 2013, 4, e12815. [Google Scholar]
- Piccioni, D.E.; Kesari, S. Clinical trials of viral therapy for malignant gliomas. Expert Rev. Anticancer Ther. 2013, 13, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, C.H.; Park, M.Y.; Park, J.S.; Park, H.M.; Sohn, H.J.; Kim, H.J.; Kim, S.G.; Oh, S.T.; Kim, T.G. Efficient co-transduction of adenoviral vectors encoding carcinoembryonic antigen and survivin into dendritic cells by the CAR-TAT adaptor molecule enhance anti-tumor immunity in a murine colorectal cancer model. Immunol. Lett. 2010, 131, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Sheng, Y.; Ren, A.; Penmatsa, S. A miR-21 hairpin structure-based gene knockdown vector. Biochem. Biophys. Res. Commun. 2010, 394, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Hu, G.; Rickles, R.J.; Hannon, G.J.; Elledge, S.J. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13212–13217. [Google Scholar] [CrossRef] [PubMed]
- Liechtenstein, T.; Perez-Janices, N.; Escors, D. Lentiviral vectors for cancer immunotherapy and clinical applications. Cancers 2013, 5, 815–837. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Kiss, A.; Schaff, Z.; Sobel, G. Claudins as diagnostic and prognostic markers in gynecological cancer. Histol. Histopathol. 2009, 24, 1607–1615. [Google Scholar] [PubMed]
- Pope, J.L.; Ahmad, R.; Bhat, A.A.; Washington, M.K.; Singh, A.B.; Dhawan, P. Claudin-1 overexpression in intestinal epithelial cells enhances susceptibility to adenamatous polyposis coli-mediated colon tumorigenesis. Mol. Cancer 2014, 13, e167. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Zou, J.; Zhang, J.; Jiang, X. DeltaNp63 promotes UMUC3 cell invasiveness and migration through claudin1 in vitro. Mol. Med. Rep. 2013, 7, 1026–1030. [Google Scholar] [PubMed]
- Iitaka, D.; Moodley, S.; Shimizu, H.; Bai, X.H.; Liu, M. PKCdelta-iPLA2-PGE2-PPARgamma signaling cascade mediates TNF-alpha induced Claudin 1 expression in human lung carcinoma cells. Cell Signal. 2015, 27, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; He, C.; Qu, Y.; Li, J.; Zhang, J.; Du, T.; Chen, X.; Yu, Y.; Liu, B.; et al. Claudin-1 enhances tumor proliferation and metastasis by regulating cell anoikis in gastric cancer. Oncotarget 2014, 6, 1652–1665. [Google Scholar]
- Qin, W.; Ren, Q.; Liu, T.; Huang, Y.; Wang, J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013, 587, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Leotlela, P.D.; Wade, M.S.; Duray, P.H.; Rhode, M.J.; Brown, H.F.; Rosenthal, D.T.; Dissanayake, S.K.; Earley, R.; Indig, F.E.; Nickoloff, B.J.; et al. Claudin-1 overexpression in melanoma is regulated by PKC and contributes to melanoma cell motility. Oncogene 2007, 26, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Sekimoto, M.; Doki, Y.; Mori, M. Expression of CLDN1 in colorectal cancer: A novel marker for prognosis. Int. J. Oncol. 2011, 39, 791–796. [Google Scholar] [PubMed]
- Blanchard, A.A.; Watson, P.H.; Shiu, R.P.; Leygue, E.; Nistor, A.; Wong, P.; Myal, Y. Differential expression of claudin 1, 3, and 4 during normal mammary gland development in the mouse. DNA Cell Biol. 2006, 25, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Morohashi, S.; Kusumi, T.; Sato, F.; Odagiri, H.; Chiba, H.; Yoshihara, S.; Hakamada, K.; Sasaki, M.; Kijima, H. Decreased expression of claudin-1 correlates with recurrence status in breast cancer. Int. J. Mol. Med. 2007, 20, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Ding, X.; Fan, Y.; Ying, J.; Zheng, S.; Lu, N.; Xu, B. A CLDN1-negative phenotype predicts poor prognosis in triple-negative breast cancer. PLoS ONE 2014, 9, e112765. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.A.; Ma, X.; Dueck, K.J.; Penner, C.; Cooper, S.C.; Mulhall, D.; Murphy, L.C.; Leygue, E.; Myal, Y. Claudin 1 expression in basal-like breast cancer is related to patient age. BMC Cancer 2013, 13, e268. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, H.; Sato, F.; Morohashi, S.; Wu, Y.; Liu, Y.; Kondo, J.; Odagiri, H.; Hakamada, K.; Kijima, H. Anti-apoptotic effect of claudin-1 in tamoxifen-treated human breast cancer MCF-7 cells. BMC Cancer 2010, 10, e548. [Google Scholar] [CrossRef] [PubMed]
- Canadas, I.; Rojo, F.; Taus, A.; Arpi, O.; Arumi-Uria, M.; Pijuan, L.; Menendez, S.; Zazo, S.; Domine, M.; Salido, M.; et al. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin. Cancer Res. 2014, 20, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Gungor, C.; Zander, H.; Effenberger, K.E.; Vashist, Y.K.; Kalinina, T.; Izbicki, J.R.; Yekebas, E.; Bockhorn, M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011, 71, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Glackin, C.A. Targeting the Twist and Wnt signaling pathways in metastatic breast cancer. Maturitas 2014, 79, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Korsching, E.; Packeisen, J.; Liedtke, C.; Hungermann, D.; Wulfing, P.; van Diest, P.J.; Brandt, B.; Boecker, W.; Buerger, H. The origin of vimentin expression in invasive breast cancer: Epithelial-mesenchymal transition, myoepithelial histogenesis or histogenesis from progenitor cells with bilinear differentiation potential? J. Pathol. 2005, 206, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, A.V.; Rooman, I.; Real, F.X. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle 2011, 10, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tian, P.; Yang, C.; Liang, Z.; Li, M.; Sims, M.; Lu, L.; Zhang, Z.; Li, H.; Pfeffer, L.M.; et al. Silencing the Double-Stranded RNA Binding Protein DGCR8 Inhibits Ovarian Cancer Cell Proliferation, Migration, and Invasion. Pharm. Res. 2015, 32, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Y.; Liu, W.; Zhao, G.; Lee, S.; Balogh, A.; Zou, Y.; Guo, Y.; Zhang, Z.; Gu, W.; et al. Doxycycline inducible Kruppel-like factor 4 lentiviral vector mediates mesenchymal to epithelial transition in ovarian cancer cells. PLoS ONE 2014, 9, e105331. [Google Scholar] [CrossRef] [PubMed]
- Myal, Y.; Leygue, E.; Blanchard, A.A. Claudin 1 in breast tumorigenesis: Revelation of a possible novel “claudin high” subset of breast cancers. J. Biomed. Biotechnol. 2010, 2010, e956897. [Google Scholar] [CrossRef] [PubMed]
- Heerma van Voss, M.R.; van Diest, P.J.; Smolders, Y.H.; Bart, J.; van der Wall, E.; van der Groep, P. Distinct claudin expression characterizes BRCA1-related breast cancer. Histopathology 2014, 65, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Singh, K.; Mangray, S.; Tavares, R.; Noble, L.; Resnick, M.B.; Yakirevich, E. Claudin expression in high-grade invasive ductal carcinoma of the breast: Correlation with the molecular subtype. Mod. Pathol. 2013, 26, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.A.; Skliris, G.P.; Watson, P.H.; Murphy, L.C.; Penner, C.; Tomes, L.; Young, T.L.; Leygue, E.; Myal, Y. Claudins 1, 3, and 4 protein expression in ER negative breast cancer correlates with markers of the basal phenotype. Virchows Arch. 2009, 454, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Di Cello, F.; Cope, L.; Li, H.; Jeschke, J.; Wang, W.; Baylin, S.B.; Zahnow, C.A. Methylation of the claudin 1 promoter is associated with loss of expression in estrogen receptor positive breast cancer. PLoS ONE 2013, 8, e68630. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Lin, X.Y.; Wang, J.; Yu, J.H.; Miao, Y.; Wang, E.H. Anti-apoptotic effect of claudin-1 on TNF-alpha-induced apoptosis in human breast cancer MCF-7 cells. Tumour Biol. 2012, 33, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Hoevel, T.; Macek, R.; Swisshelm, K.; Kubbies, M. Reexpression of the TJ protein CLDN1 induces apoptosis in breast tumor spheroids. Int. J. Cancer 2004, 108, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Estrada, O.M.; Culleres, A.; Soriano, F.X.; Peinado, H.; Bolos, V.; Martinez, F.O.; Reina, M.; Cano, A.; Fabre, M.; Vilaro, S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem. J. 2006, 394, 449–457. [Google Scholar] [PubMed]
- Van Pham, P.; Vu, N.B.; Duong, T.T.; Nguyen, T.T.; Truong, N.H.; Phan, N.L.; Vuong, T.G.; Pham, V.Q.; Nguyen, H.M.; Nguyen, K.T.; et al. Suppression of human breast tumors in NOD/SCID mice by CD44 shRNA gene therapy combined with doxorubicin treatment. OncoTargets Ther. 2012, 5, 77–84. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zou, Y.; Gu, Q.; Zhao, G.; Gray, H.; Pfeffer, L.M.; Yue, J. Lentiviral Vector Mediated Claudin1 Silencing Inhibits Epithelial to Mesenchymal Transition in Breast Cancer Cells. Viruses 2015, 7, 2965-2979. https://doi.org/10.3390/v7062755
Zhao X, Zou Y, Gu Q, Zhao G, Gray H, Pfeffer LM, Yue J. Lentiviral Vector Mediated Claudin1 Silencing Inhibits Epithelial to Mesenchymal Transition in Breast Cancer Cells. Viruses. 2015; 7(6):2965-2979. https://doi.org/10.3390/v7062755
Chicago/Turabian StyleZhao, Xianqi, Yanan Zou, Qingqing Gu, Guannan Zhao, Horace Gray, Lawrence M. Pfeffer, and Junming Yue. 2015. "Lentiviral Vector Mediated Claudin1 Silencing Inhibits Epithelial to Mesenchymal Transition in Breast Cancer Cells" Viruses 7, no. 6: 2965-2979. https://doi.org/10.3390/v7062755
APA StyleZhao, X., Zou, Y., Gu, Q., Zhao, G., Gray, H., Pfeffer, L. M., & Yue, J. (2015). Lentiviral Vector Mediated Claudin1 Silencing Inhibits Epithelial to Mesenchymal Transition in Breast Cancer Cells. Viruses, 7(6), 2965-2979. https://doi.org/10.3390/v7062755