
Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
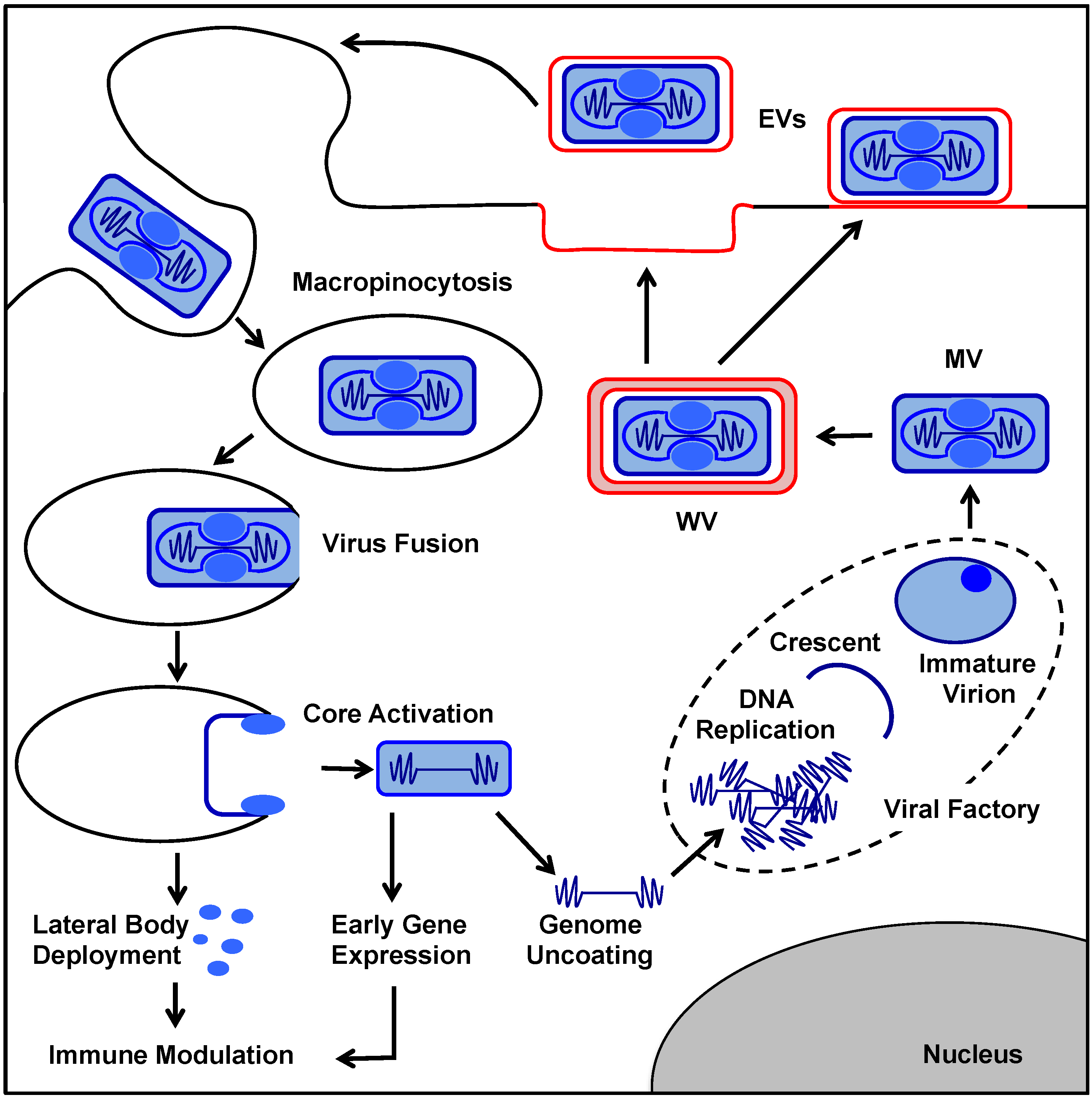
2. VACV Replication Cycle

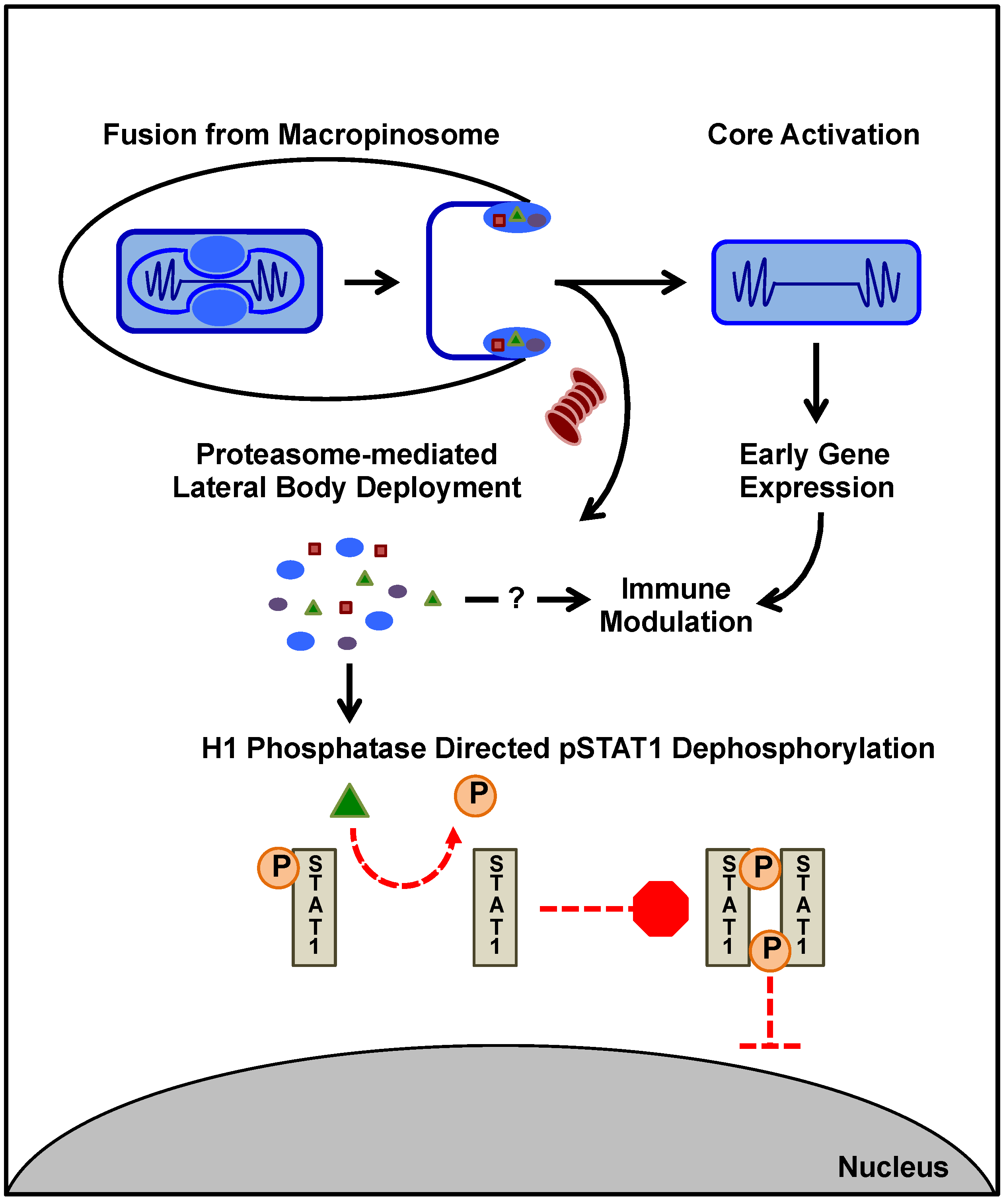
3. Immune Suppression during MV Entry
4. Post Entry VACV Immunomodulation

5. VACV EV Formation
5.1. Overview: From MV to EV

5.2. WV Formation
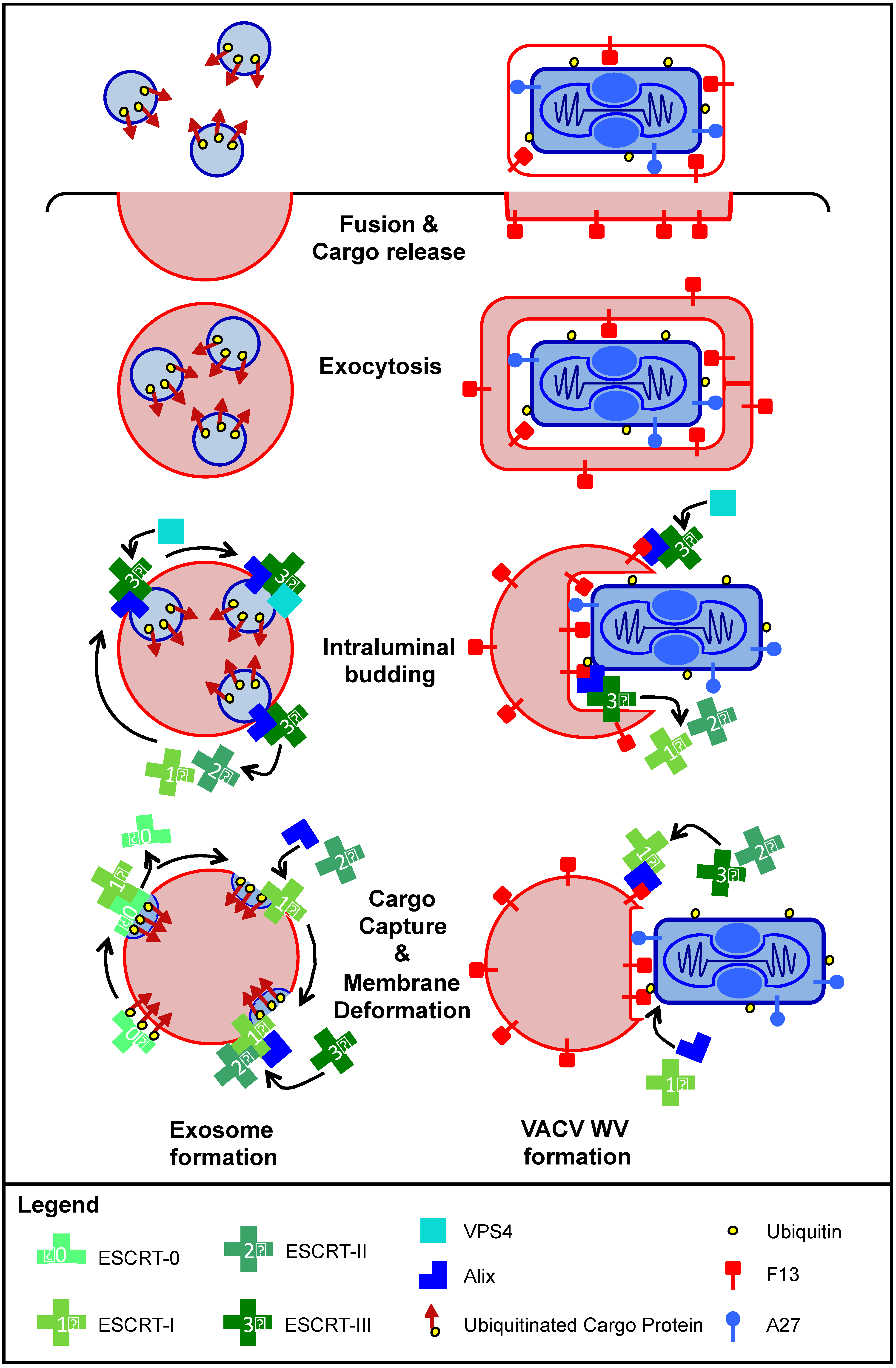
5.3. Is EV Formation an Exosome-Like Process?

6. Immune Evasion Role of the EV Membrane
7. Perspectives
Acknowledgments
Conflicts of Interest
References
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.C.; Jacques, D.A. Intracellular immunity: Finding the enemy within—How cells recognize and respond to intracellular pathogens. J. Leukoc. Biol. 2014, 96, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; MacMicking, J.D.; James, L.C. Cellular self-defense: How cell-autonomous immunity protects against pathogens. Science 2013, 340, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Beyaert, R. Role of toll-like receptors in pathogen recognition. Clin. Microbiol. Rev. 2003, 16, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.C.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular sensing of complement C3 activates cell autonomous immunity. Science 2014, 345, 1256070. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Kagan, J.C. Intracellular pathogen detection by Rig-I-like receptors. Adv. Immunol. 2013, 117, 99–125. [Google Scholar] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479C–480C, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef] [PubMed]
- Knipe, S.; Howley, P. Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Kelly, B.J.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 2009, 145, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 2013, 4, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, K.S.; Flano, E.; Wu, T.T.; Tong, L.M.; Park, A.N.; Song, M.J.; Sanchez, D.J.; O’Connell, R.M.; Cheng, G.; et al. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 2009, 5, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Trgovcich, J.; Johnson, D.; Roizman, B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J. Virol. 2002, 76, 6974–6986. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. General Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F.; Wittek, R.; Dumbell, K. The Orthopoxviruses; Academic Press: London, UK, 1989. [Google Scholar]
- Fenner, F.; Anderson, D.; Arita, I.; Jezek, Z.; Ladnyi, D. Smallpox and Its Eradication; World Health Organization: Geneva, Switzerland, 1988. [Google Scholar]
- Hollinshead, M.; Vanderplasschen, A.; Smith, G.L.; Vaux, D.J. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 1999, 73, 1503–1517. [Google Scholar] [PubMed]
- Cyrklaff, M.; Risco, C.; Fernandez, J.J.; Jimenez, M.V.; Esteban, M.; Baumeister, W.; Carrascosa, J.L. Cryo-electron tomography of vaccinia virus. Proc. Natl. Acad. Sci. USA 2005, 102, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Ulaeto, D.; Grosenbach, D.; Hruby, D.E. The vaccinia virus 4c and A-type inclusion proteins are specific markers for the intracellular mature virus particle. J. Virol. 1996, 70, 3372–3377. [Google Scholar] [PubMed]
- Smith, G.L.; Law, M. The exit of vaccinia virus from infected cells. Virus Res. 2004, 106, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Murphy, B.J.; Law, M. Vaccinia virus motility. Annu. Rev. Microbiol. 2003, 57, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.G. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J. General Virol. 1980, 50, 89–100. [Google Scholar] [CrossRef]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.; Helenius, A.; Mercer, J. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 2011, 30, 3647–3661. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.; Mercer, J. Poxvirus host cell entry. Curr. Opin. Virol. 2012, 2, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Lu, T.Y.; Bair, C.H.; Chang, Y.S.; Jwo, J.K.; Chang, W. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into Hela cells through fluid phase endocytosis. J. Virol. 2008, 82, 7988–7999. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, K.J.; Wilkinson, J.; Miranda-Saksena, M.; McInerney, G.M.; Byth-Wilson, K.; Robinson, P.J.; Cunningham, A.L. A differential role for macropinocytosis in mediating entry of the two forms of vaccinia virus into dendritic cells. PLoS Pathog. 2010, 6, e1000866. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Gulping rather than sipping: Macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 2012, 15, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus cell entry: How many proteins does it take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Townsley, A.C.; Weisberg, A.S.; Wagenaar, T.R.; Moss, B. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 2006, 80, 8899–8908. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology 1996, 217, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Townsley, A.C.; Moss, B. Two distinct low-pH steps promote entry of vaccinia virus. J. Virol. 2007, 81, 8613–8620. [Google Scholar] [CrossRef] [PubMed]
- Rizopoulos, Z.; Balistreri, G.; Kilcher, S.; Martin, C.K.; Syedbasha, M.; Helenius, A.; Mercer, J. Vaccinia virus infection requires maturation of macropinosomes. Traffic 2015, 16, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Chang, Y.X.; Izmailyan, R.; Tang, Y.L.; Chang, W. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into Hela, CHO-K1, and l cells. J. Virol. 2010, 84, 8422–8432. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Moss, B. Interaction of the vaccinia virus RNA polymerase-associated 94-kilodalton protein with the early transcription factor. J. Virol. 2009, 83, 12018–12026. [Google Scholar] [CrossRef] [PubMed]
- Dales, S. The uptake and development of vaccinia virus in strain l cells followed with labeled viral deoxyribonucleic acid. J. Cell Biol. 1963, 18, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.; Snijder, E.J.; Schleich, S.; Roos, N.; Griffiths, G.; Locker, J.K. Characterization of vaccinia virus intracellular cores: Implications for viral uncoating and core structure. J. Virol. 2000, 74, 3525–3536. [Google Scholar] [CrossRef] [PubMed]
- Locker, J.K.; Griffiths, G. An unconventional role for cytoplasmic disulfide bonds in vaccinia virus proteins. J. Cell Biol. 1999, 144, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Schmidt, F.I.; Schneider, C.; Kopf, M.; Helenius, A.; Mercer, J. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 2014, 15, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C.; Moussatche, N.; Traktman, P. In a nutshell: Structure and assembly of the vaccinia virion. Adv. Virus Res. 2006, 66, 31–124. [Google Scholar] [PubMed]
- Liu, L.; Cooper, T.; Howley, P.M.; Hayball, J.D. From crescent to mature virion: Vaccinia virus assembly and maturation. Viruses 2014, 6, 3787–3808. [Google Scholar] [CrossRef] [PubMed]
- Doceul, V.; Hollinshead, M.; Breiman, A.; Laval, K.; Smith, G.L. Protein B5 is required on extracellular enveloped vaccinia virus for repulsion of superinfecting virions. J. General Virol. 2012, 93, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, M.; Sodeik, B.; Ericsson, M.; Wolffe, E.J.; Shida, H.; Hiller, G.; Griffiths, G. Assembly of vaccinia virus: The second wrapping cisterna is derived from the trans Golgi network. J. Virol. 1994, 68, 130–147. [Google Scholar] [PubMed]
- Hiller, G.; Weber, K. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 1985, 55, 651–659. [Google Scholar] [PubMed]
- Sodeik, B.; Doms, R.W.; Ericsson, M.; Hiller, G.; Machamer, C.E.; van’t Hof, W.; van Meer, G.; Moss, B.; Griffiths, G. Assembly of vaccinia virus: Role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 1993, 121, 521–541. [Google Scholar] [CrossRef] [PubMed]
- Tooze, J.; Hollinshead, M.; Reis, B.; Radsak, K.; Kern, H. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 1993, 60, 163–178. [Google Scholar] [PubMed]
- Van Eijl, H.; Hollinshead, M.; Rodger, G.; Zhang, W.H.; Smith, G.L. The vaccinia virus F12l protein is associated with intracellular enveloped virus particles and is required for their egress to the cell surface. J. General Virol. 2002, 83, 195–207. [Google Scholar]
- Leite, F.; Way, M. The role of signalling and the cytoskeleton during vaccinia virus egress. Virus Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 2001, 75, 11651–11663. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J. Virol. 2001, 75, 4802–4813. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Apoptotic mimicry: Phosphatidylserine-mediated macropinocytosis of vaccinia virus. Ann. N. Y. Acad. Sci. 2010, 1209, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Elkon, K.B.; Ma, X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity 2004, 21, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Cvetanovic, M.; Ucker, D.S. Innate immune discrimination of apoptotic cells: Repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 2004, 172, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Vanlandschoot, P.; Leroux-Roels, G. Viral apoptotic mimicry: An immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 2003, 24, 144–147. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Zagorska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Knebel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. USA 2010, 107, 9346–9351. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, J.P.; Moss, B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. USA 2009, 106, 17517–17521. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.P.; Jeon, O.Y.; Kilcher, S.; Moest, H.; Henning, L.M.; Jost, C.; Pluckthun, A.; Mercer, J.; Aebersold, R.; Carreira, E.M.; et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nat. Biotechnol. 2012, 30, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Z.; Fuhlbrigge, R.C.; Pena-Cruz, V.; Lieberman, J.; Kupper, T.S. Vaccinia virus induces strong immunoregulatory cytokine production in healthy human epidermal keratinocytes: A novel strategy for immune evasion. J. Virol. 2005, 79, 7363–7370. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, D.; Ennis, F.A.; Terajima, M. Pathogeneses of respiratory infections with virulent and attenuated vaccinia viruses. Virol. J. 2007, 4, e22. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Franz, K.M.; Kagan, J.C. PRRs are watching you: Localization of innate sensing and signalling regulators. Virology 2015, 479C–480C, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Greber, U.F. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol. 2013, 21, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Peters, D. Morphology of resting vaccinia virus. Nature 1956, 178, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, Y.; Oie, M.; Tsuruhara, T. Location of DNA-binding proteins and disulfide-linked proteins in vaccinia virus structural elements. J. Virol. 1984, 50, 929–938. [Google Scholar] [PubMed]
- Chung, C.S.; Chen, C.H.; Ho, M.Y.; Huang, C.Y.; Liao, C.L.; Chang, W. Vaccinia virus proteome: Identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 2006, 80, 2127–2140. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Najarro, P.; Traktman, P.; Lewis, J.A. Vaccinia virus blocks gamma interferon signal transduction: Viral VH1 phosphatase reverses Stat1 activation. J. Virol. 2001, 75, 3185–3196. [Google Scholar] [CrossRef] [PubMed]
- Sarov, I.; Joklik, W.K. Studies on the nature and location of the capsid polypeptides of vaccinia virions. Virology 1972, 50, 579–592. [Google Scholar] [CrossRef]
- Wickramasekera, N.T.; Traktman, P. Structure/function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J. Virol. 2010, 84, 6846–6860. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.A.; Silva, P.N.; Pereira, A.C.; de Sousa, L.P.; Ferreira, P.C.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Weisberg, A.S.; Moss, B. A glutaredoxin, encoded by the G4L gene of vaccinia virus, is essential for virion morphogenesis. J. Virol. 2000, 74, 9175–9183. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lemon, B.; Traktman, P. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J. Virol. 1995, 69, 7823–7834. [Google Scholar] [PubMed]
- Vanderplasschen, A.; Mathew, E.; Hollinshead, M.; Sim, R.B.; Smith, G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. USA 1998, 95, 7544–7549. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Carter, G.C.; Roberts, K.L.; Hollinshead, M.; Smith, G.L. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 2006, 103, 5989–5994. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.M.; Hollinshead, M.; Smith, G.L. The vaccinia virus A27l protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J. General Virol. 2000, 81, 47–58. [Google Scholar]
- Ward, B.M. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 2005, 79, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Roper, R.L.; Payne, L.G.; Moss, B. Extracellular vaccinia virus envelope glycoprotein encoded by the A33R gene. J. Virol. 1996, 70, 3753–3762. [Google Scholar] [PubMed]
- Duncan, S.A.; Smith, G.L. Identification and characterization of an extracellular envelope glycoprotein affecting vaccinia virus egress. J. Virol. 1992, 66, 1610–1621. [Google Scholar] [PubMed]
- Parkinson, J.E.; Smith, G.L. Vaccinia virus gene A36R encodes a M(r) 43–50 K protein on the surface of extracellular enveloped virus. Virology 1994, 204, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Shida, H. Nucleotide sequence of the vaccinia virus hemagglutinin gene. Virology 1986, 150, 451–462. [Google Scholar] [CrossRef]
- Zhang, W.H.; Wilcock, D.; Smith, G.L. Vaccinia virus F12l protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 2000, 74, 11654–11662. [Google Scholar] [CrossRef] [PubMed]
- Hirt, P.; Hiller, G.; Wittek, R. Localization and fine structure of a vaccinia virus gene encoding an envelope antigen. J. Virol. 1986, 58, 757–764. [Google Scholar] [PubMed]
- Engelstad, M.; Smith, G.L. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 1993, 194, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Domi, A.; Weisberg, A.S.; Moss, B. Vaccinia virus E2L null mutants exhibit a major reduction in extracellular virion formation and virus spread. J. Virol. 2008, 82, 4215–4226. [Google Scholar] [CrossRef] [PubMed]
- Brum, L.M.; Turner, P.C.; Devick, H.; Baquero, M.T.; Moyer, R.W. Plasma membrane localization and fusion inhibitory activity of the cowpox virus serpin SPI-3 require a functional signal sequence and the virus encoded hemagglutinin. Virology 2003, 306, 289–302. [Google Scholar] [CrossRef]
- Wagenaar, T.R.; Moss, B. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 2007, 81, 6286–6293. [Google Scholar] [CrossRef] [PubMed]
- Ulaeto, D.; Grosenbach, D.; Hruby, D.E. Brefeldin-A inhibits vaccinia virus envelopment but does not prevent normal processing and localization of the putative envelopment receptor P37. J. General Virol. 1995, 76, 103–111. [Google Scholar] [CrossRef]
- Husain, M.; Moss, B. Similarities in the induction of post-Golgi vesicles by the vaccinia virus F13l protein and phospholipase D. J. Virol. 2002, 76, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Moss, B. Vaccinia virus F13l protein with a conserved phospholipase catalytic motif induces colocalization of the B5R envelope glycoprotein in post-Golgi vesicles. J. Virol. 2001, 75, 7528–7542. [Google Scholar] [CrossRef] [PubMed]
- Beard, P.M.; Griffiths, S.J.; Gonzalez, O.; Haga, I.R.; Jowers, T.P.; Reynolds, D.K.; Wildenhain, J.; Tekotte, H.; Auer, M.; Tyers, M.; et al. A loss of function analysis of host factors influencing vaccinia virus replication by RNA interference. PLoS ONE 2014, 9, e98431. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Moss, B. Role of receptor-mediated endocytosis in the formation of vaccinia virus extracellular enveloped particles. J. Virol. 2005, 79, 4080–4089. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.F.; Smith, G.L. IPTG-dependent vaccinia virus: Identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 1990, 18, 5347–5351. [Google Scholar] [CrossRef] [PubMed]
- Blasco, R.; Moss, B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 1991, 65, 5910–5920. [Google Scholar] [PubMed]
- Wolffe, E.J.; Isaacs, S.N.; Moss, B. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 1993, 67, 4732–4741. [Google Scholar] [PubMed]
- Grosenbach, D.W.; Ulaeto, D.O.; Hruby, D.E. Palmitylation of the vaccinia virus 37-kDa major envelope antigen. Identification of a conserved acceptor motif and biological relevance. J. Biol. Chem. 1997, 272, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Weisberg, A.; Moss, B. Topology of epitope-tagged F13l protein, a major membrane component of extracellular vaccinia virions. Virology 2003, 308, 233–242. [Google Scholar] [CrossRef]
- Roper, R.L.; Moss, B. Envelope formation is blocked by mutation of a sequence related to the HKD phospholipid metabolism motif in the vaccinia virus F13l protein. J. Virol. 1999, 73, 1108–1117. [Google Scholar] [PubMed]
- Baek, S.H.; Kwak, J.Y.; Lee, S.H.; Lee, T.; Ryu, S.H.; Uhlinger, D.J.; Lambeth, J.D. Lipase activities of p37, the major envelope protein of vaccinia virus. J. Biol. Chem. 1997, 272, 32042–32049. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Moss, B. Intracellular trafficking of a palmitoylated membrane-associated protein component of enveloped vaccinia virus. J. Virol. 2003, 77, 9008–9019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Honeychurch, K.M.; Yang, G.; Byrd, C.M.; Harver, C.; Hruby, D.E.; Jordan, R. Vaccinia virus p37 interacts with host proteins associated with Le-derived transport vesicle biogenesis. Virol. J. 2009, 6, e44. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.S.; Hanna, J.; Simon, I.; Krise, J.; Barbero, P.; Pfeffer, S.R. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science 2001, 292, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.; Pfeffer, S.R. TIP47: A cargo selection device for mannose 6-phosphate receptor trafficking. Cell 1998, 93, 433–443. [Google Scholar] [CrossRef]
- Ploen, D.; Hafirassou, M.L.; Himmelsbach, K.; Schille, S.A.; Biniossek, M.L.; Baumert, T.F.; Schuster, C.; Hildt, E. TIP47 is associated with the hepatitis C virus and its interaction with Rab9 is required for release of viral particles. Eur. J. Cell Biol. 2013, 92, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.L.; Mavrakis, M.; McDonald, N.J.; Yilla, M.; Sheng, J.; Bellini, W.J.; Zhao, L.; Le Doux, J.M.; Shaw, M.W.; Luo, C.C.; et al. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J. Virol. 2005, 79, 11742–11751. [Google Scholar] [CrossRef] [PubMed]
- Blot, G.; Janvier, K.; Le Panse, S.; Benarous, R.; Berlioz-Torrent, C. Targeting of the human immunodeficiency virus type 1 envelope to the trans-Golgi network through binding to TIP47 is required for env incorporation into virions and infectivity. J. Virol. 2003, 77, 6931–6945. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.M.; Yang, G.; Jordan, R.; Hruby, D.E. The vaccinia virus F13l YPPL motif is required for efficient release of extracellular enveloped virus. J. Virol. 2007, 81, 7310–7315. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Lamb, R.A. Mechanisms for enveloped virus budding: Can some viruses do without an ESCRT? Virology 2008, 372, 221–232. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. Aip1/alix is a binding partner for hiv-1 p6 and eiav p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.H.; Mayer, R.J.; Dixon, L.K. A lipid modified ubiquitin is packaged into particles of several enveloped viruses. FEBS Lett. 1999, 444, 136–139. [Google Scholar] [CrossRef]
- Krauss, O.; Hollinshead, R.; Hollinshead, M.; Smith, G.L. An investigation of incorporation of cellular antigens into vaccinia virus particles. J. General Virol. 2002, 83, 2347–2359. [Google Scholar]
- Ward, B.M.; Moss, B. Golgi network targeting and plasma membrane internalization signals in vaccinia virus B5R envelope protein. J. Virol. 2000, 74, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- Amanna, I.J.; Slifka, M.K.; Crotty, S. Immunity and immunological memory following smallpox vaccination. Immunol. Rev. 2006, 211, 320–337. [Google Scholar] [CrossRef] [PubMed]
- Edghill-Smith, Y.; Golding, H.; Manischewitz, J.; King, L.R.; Scott, D.; Bray, M.; Nalca, A.; Hooper, J.W.; Whitehouse, C.A.; Schmitz, J.E.; et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 2005, 11, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Boulter, E.A.; Appleyard, G. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog. Med. Virol. 1973, 16, 86–108. [Google Scholar] [PubMed]
- Law, M.; Putz, M.M.; Smith, G.L. An investigation of the therapeutic value of vaccinia-immune IgG in a mouse pneumonia model. J. General Virol. 2005, 86, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Duke-Cohan, J.S.; Wollenick, K.; Witten, E.A.; Seaman, M.S.; Baden, L.R.; Dolin, R.; Reinherz, E.L. The heterogeneity of human antibody responses to vaccinia virus revealed through use of focused protein arrays. Vaccine 2009, 27, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.M.; Midgley, C.M.; Law, M.; Smith, G.L. Quantification of antibody responses against multiple antigens of the two infectious forms of vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 2006, 12, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. General Virol. 2002, 83, 2915–2931. [Google Scholar]
- Mercer, J.; Traktman, P. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 2003, 77, 8857–8871. [Google Scholar] [CrossRef] [PubMed]
- Machiels, B.; Lete, C.; Guillaume, A.; Mast, J.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. Antibody evasion by a gammaherpesvirus O-glycan shield. PLoS Pathog. 2011, 7, e1002387. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.E.; Xiao, Y.; Eisenberg, R.J.; Cohen, G.H.; Isaacs, S.N. Antibody against extracellular vaccinia virus (EV) protects mice through complement and Fc receptors. PLoS ONE 2011, 6, e20597. [Google Scholar] [CrossRef] [PubMed]
- Benhnia, M.R.; McCausland, M.M.; Moyron, J.; Laudenslager, J.; Granger, S.; Rickert, S.; Koriazova, L.; Kubo, R.; Kato, S.; Crotty, S. Vaccinia virus extracellular enveloped virion neutralization in vitro and protection in vivo depend on complement. J. Virol. 2009, 83, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Girgis, N.M.; Dehaven, B.C.; Fan, X.; Viner, K.M.; Shamim, M.; Isaacs, S.N. Cell surface expression of the vaccinia virus complement control protein is mediated by interaction with the viral A56 protein and protects infected cells from complement attack. J. Virol. 2008, 82, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Smith, G.L. Antibody neutralization of the extracellular enveloped form of vaccinia virus. Virology 2001, 280, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the dead: Apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.M.; King, S.W.; Thorpe, P.E. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 2008, 14, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; McFadden, G. Oncolytic poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. A cutting-edge view on the current state of antiviral drug development. Med. Res. Rev. 2013, 33, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidgood, S.R.; Mercer, J. Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses. Viruses 2015, 7, 4800-4825. https://doi.org/10.3390/v7082844
Bidgood SR, Mercer J. Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses. Viruses. 2015; 7(8):4800-4825. https://doi.org/10.3390/v7082844
Chicago/Turabian StyleBidgood, Susanna R., and Jason Mercer. 2015. "Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses" Viruses 7, no. 8: 4800-4825. https://doi.org/10.3390/v7082844
APA StyleBidgood, S. R., & Mercer, J. (2015). Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses. Viruses, 7(8), 4800-4825. https://doi.org/10.3390/v7082844