
Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Routine Subculture
2.2. Studying the Viral Cycle Using TEM
2.3. Genome Study
2.4. Accession Number
3. Results
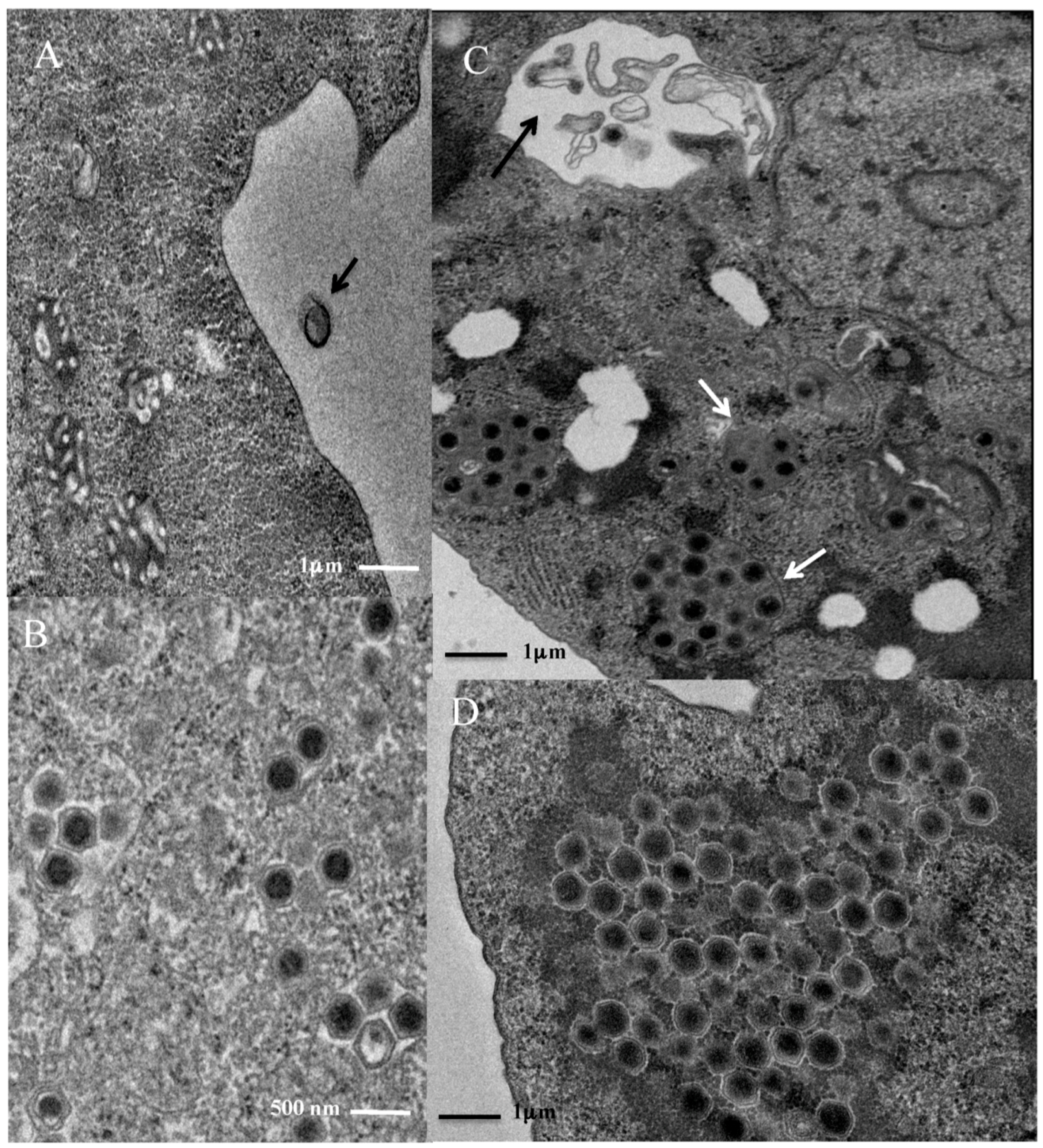
3.1. Replication Cycle
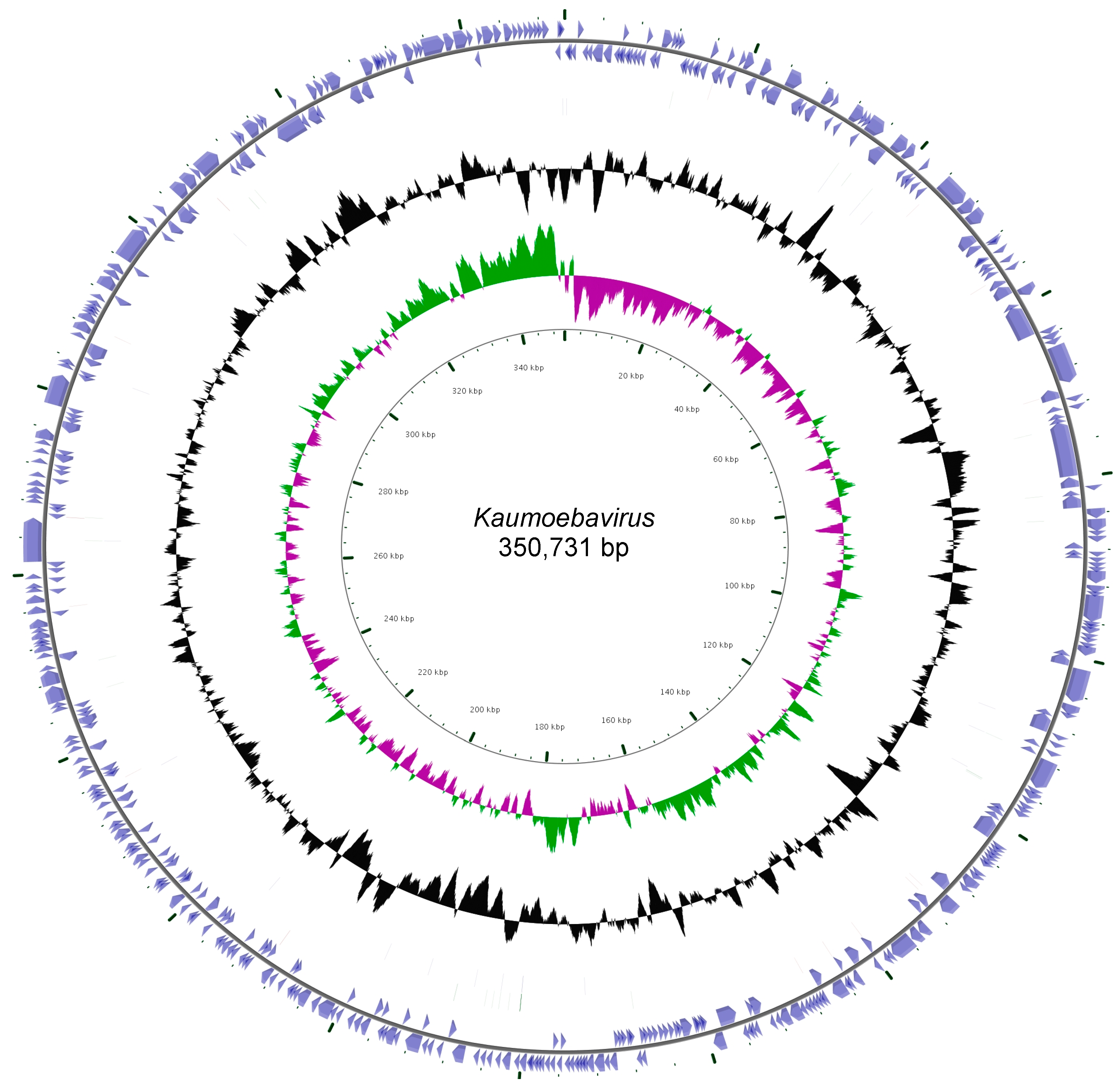
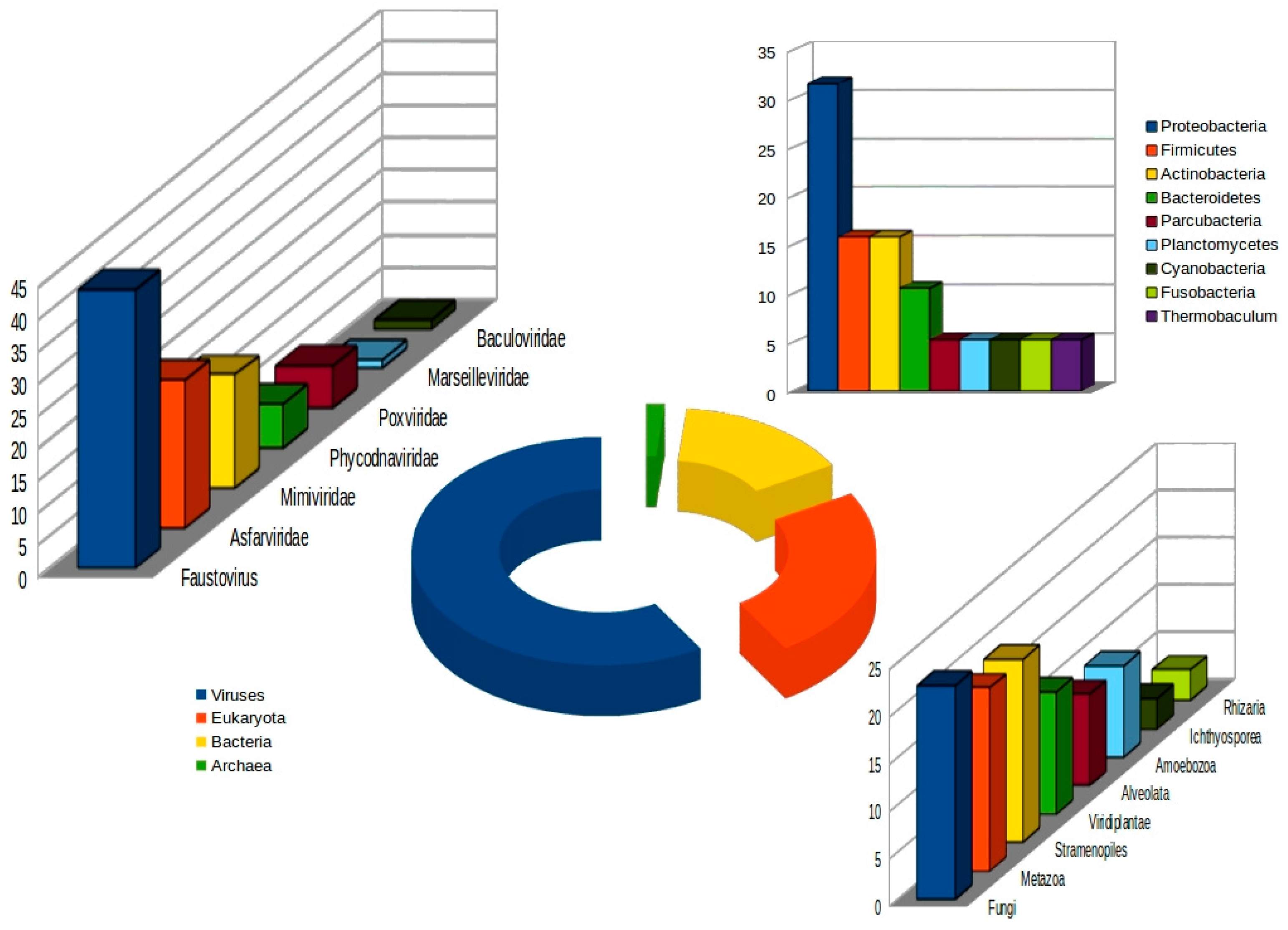
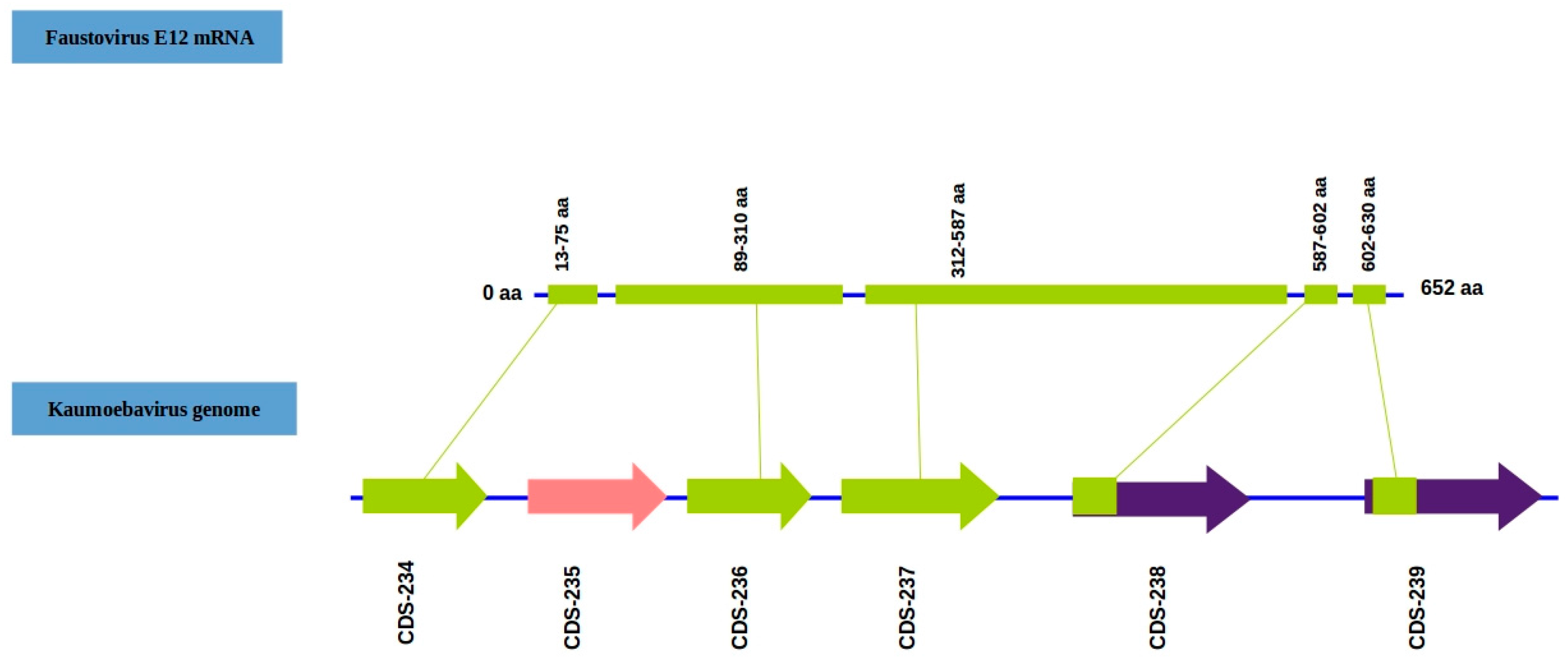
3.2. Genomic Features
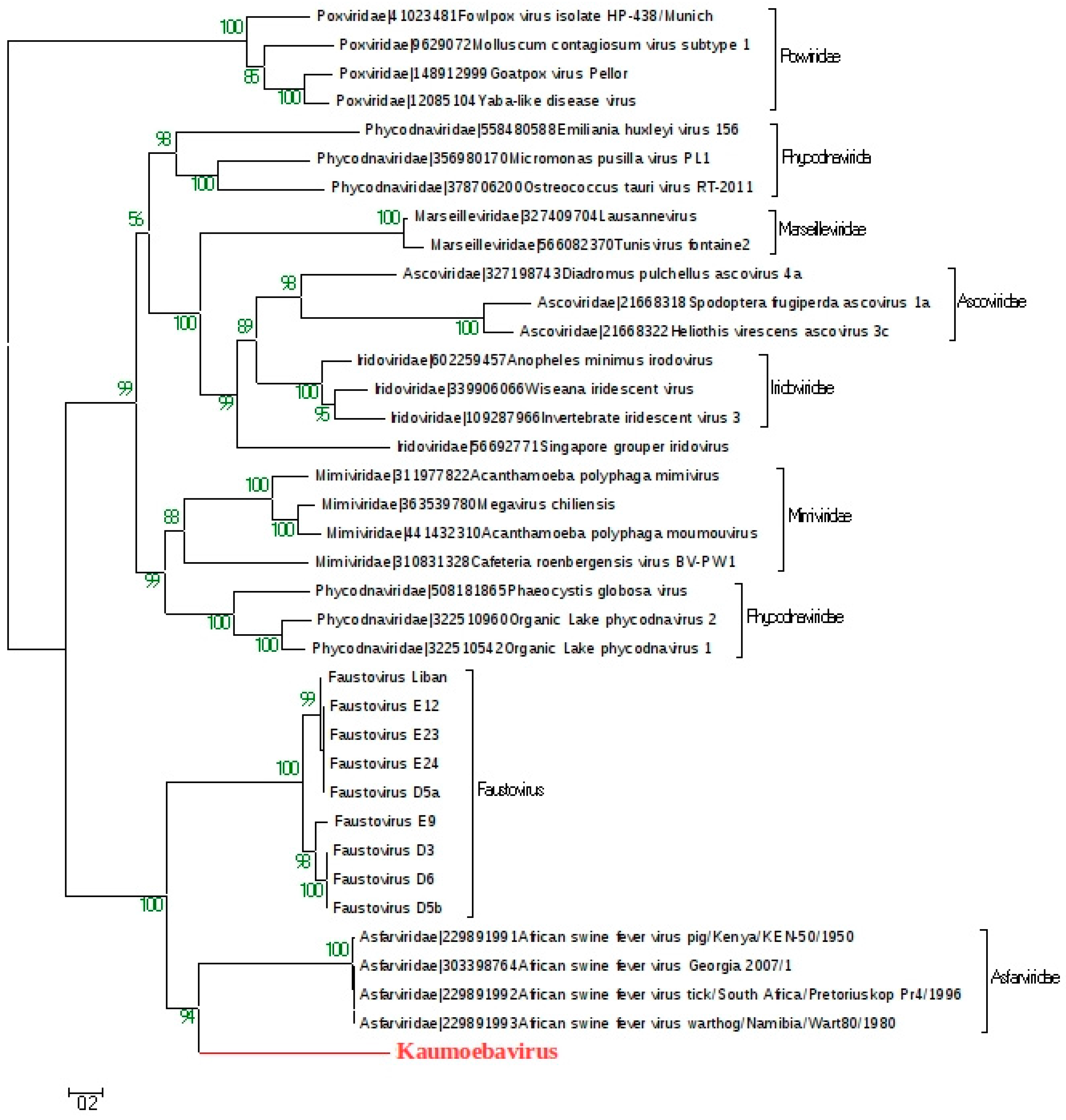
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; De Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant Mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed]
- Yoosuf, N.; Yutin, N.; Colson, P.; Shabalina, S.A.; Pagnier, I.; Robert, C.; Azza, S.; Klose, T.; Wong, J.; Rossmann, M.G.; et al. Related giant viruses in distant locations and different habitats: Acanthamoeba polyphaga moumouvirus represents a third lineage of the Mimiviridae that is close to the megavirus lineage. Genome Biol. Evol. 2012, 4, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Saadi, H.; Pagnier, I.; Colson, P.; Cherif, J.K.; Beji, M.; Boughalmi, M.; Azza, S.; Armstrong, N.; Robert, C.; Fournous, G.; et al. First isolation of Mimivirus in a patient with pneumonia. Clin. Infect. Dis. 2013, 57, e127–e134. [Google Scholar] [CrossRef] [PubMed]
- Saadi, H.; Reteno, D.G.; Colson, P.; Aherfi, S.; Minodier, P.; Pagnier, I.; Raoult, D.; La Scola, B. Shan virus: A new mimivirus isolated from the stool of a Tunisian patient with pneumonia. Intervirology 2013, 56, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Boughalmi, M.; Pagnier, I.; Aherfi, S.; Colson, P.; Raoult, D.; La Scola, B. First isolation of a Marseillevirus in the Diptera Syrphidae Eristalis tenax. Intervirology 2013, 56, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Boughalmi, M.; Saadi, H.; Pagnier, I.; Colson, P.; Fournous, G.; Raoult, D.; La Scola, B. High-throughput isolation of giant viruses of the Mimiviridae and Marseilleviridae families in the Tunisian environment. Environ. Microbiol. 2013, 15, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Aherfi, S.; Boughalmi, M.; Pagnier, I.; Fournous, G.; La Scola, B.; Raoult, D.; Colson, P. Complete genome sequence of tunisvirus, a new member of the proposed family Marseilleviridae. Arch. Virol. 2014, 159, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Coute, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempica, J.M.; Ramusb, C.; Bruleyb, C.; Labadiee, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, E5327–E5335. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; De Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Bou, K.J.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6589. [Google Scholar] [CrossRef] [PubMed]
- Benamar, S.; Reteno, D.G.; Labas, N.; Raoult, D.; La Scola, B.; Bandaly, V.; Martinez-Salas, E.; Shuang-Yong, X. Faustoviruses: Comparative genomics of new Megavirales family members. Front. Microbiol. 2016, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khalil, J.Y.B.; Andreani, J.; La Scola, B. Updating strategies for isolating and discovering giant viruses. Curr. Opin. Microbiol. 2016, 31, 80–87. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Campocasso, A.; N’dong, R.; Fournous, G.; Barrassi, L.; Flaudrops, C.; Raoult, D. Tentative characterization of new environmental giant viruses by MALDI-TOF mass spectrometry. Intervirology 2010, 53, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2011, 13 (Suppl. S14), S8. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program for the detection of transfer RNA and transfer-messenger RNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Kannan, L.; Coleman, M.K.; Wolf, Y.I.; Sorokin, A.; Koonin, E.V.; Mushegian, A. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 2010, 26, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; De la Concha-Bermejillo, A.; Lehmkuhl, H.D.; Piccone, M.E.; Kutish, G.F.; Rock, D.L. Genomes of the Parapoxviruses orf virus and bovine papular stomatitis virus. J. Virol. 2004, 78, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Klose, T.; Reteno, D.G.; Benamar, S.; Hollerbach, A.; Colson, P.; La Scola, B.; Rossmann, M.G. Structure of faustovirus, a large dsDNA virus. Proc. Natl. Acad. Sci. USA 2016, 113, 6206–6211. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajrai, L.H.; Benamar, S.; Azhar, E.I.; Robert, C.; Levasseur, A.; Raoult, D.; La Scola, B. Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae. Viruses 2016, 8, 278. https://doi.org/10.3390/v8110278
Bajrai LH, Benamar S, Azhar EI, Robert C, Levasseur A, Raoult D, La Scola B. Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae. Viruses. 2016; 8(11):278. https://doi.org/10.3390/v8110278
Chicago/Turabian StyleBajrai, Leena H., Samia Benamar, Esam I. Azhar, Catherine Robert, Anthony Levasseur, Didier Raoult, and Bernard La Scola. 2016. "Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae" Viruses 8, no. 11: 278. https://doi.org/10.3390/v8110278
APA StyleBajrai, L. H., Benamar, S., Azhar, E. I., Robert, C., Levasseur, A., Raoult, D., & La Scola, B. (2016). Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae. Viruses, 8(11), 278. https://doi.org/10.3390/v8110278