
Host–Pathogen Interactions in Measles Virus Replication and Anti-Viral Immunity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
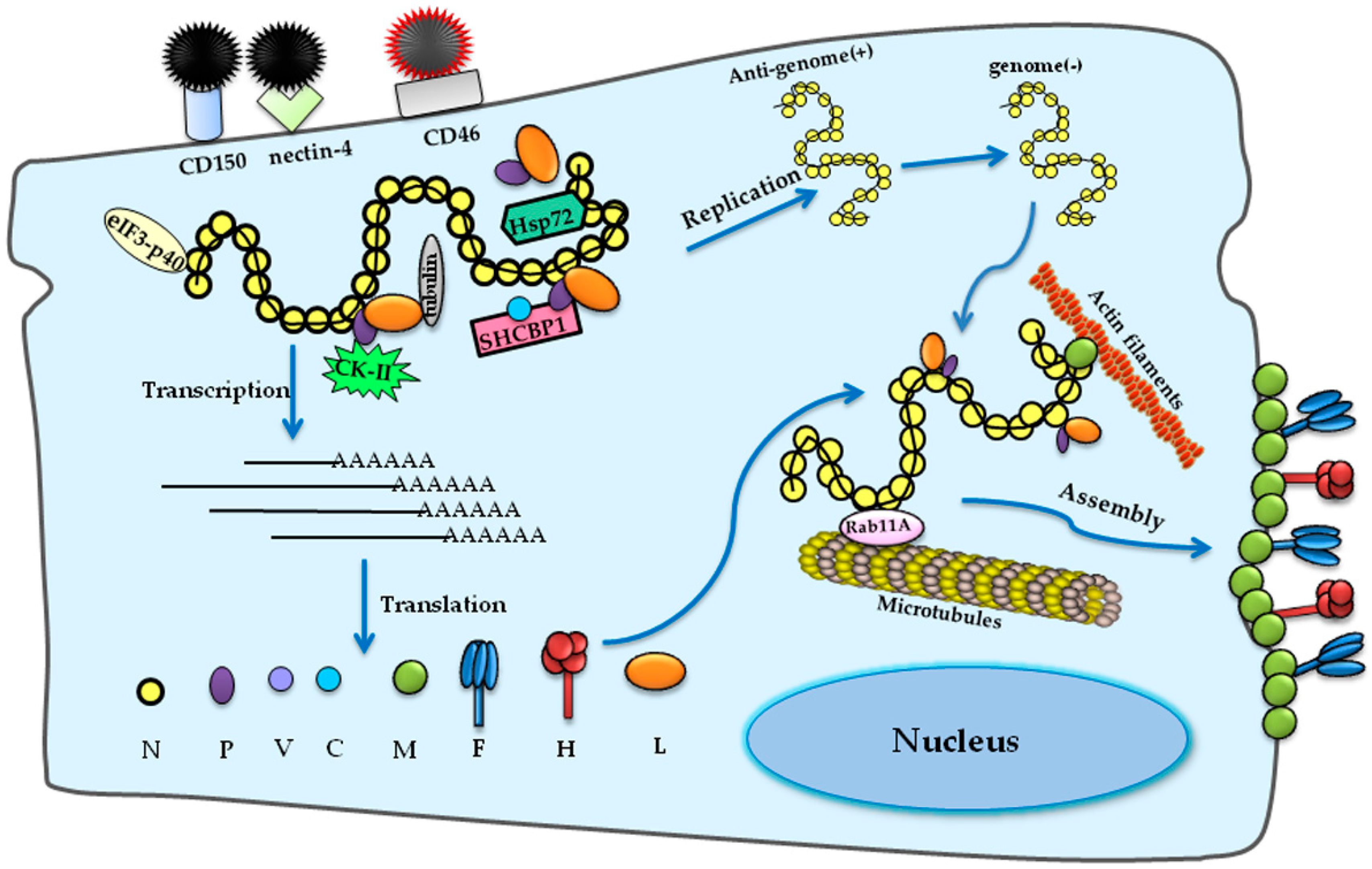
2. Genome and Lifecycle
2.1. Genome
2.2. Cell Entry
2.3. Transcription and Replication
2.4. Assembly and Egress
3. Interaction between MeV and Cellular Factors
3.1. Host Factors Involved in MeV Replication
3.1.1. Host Factors Involved in MeV Entry
3.1.2. Host Factors Involved in MeV RNA Synthesis and Assembly
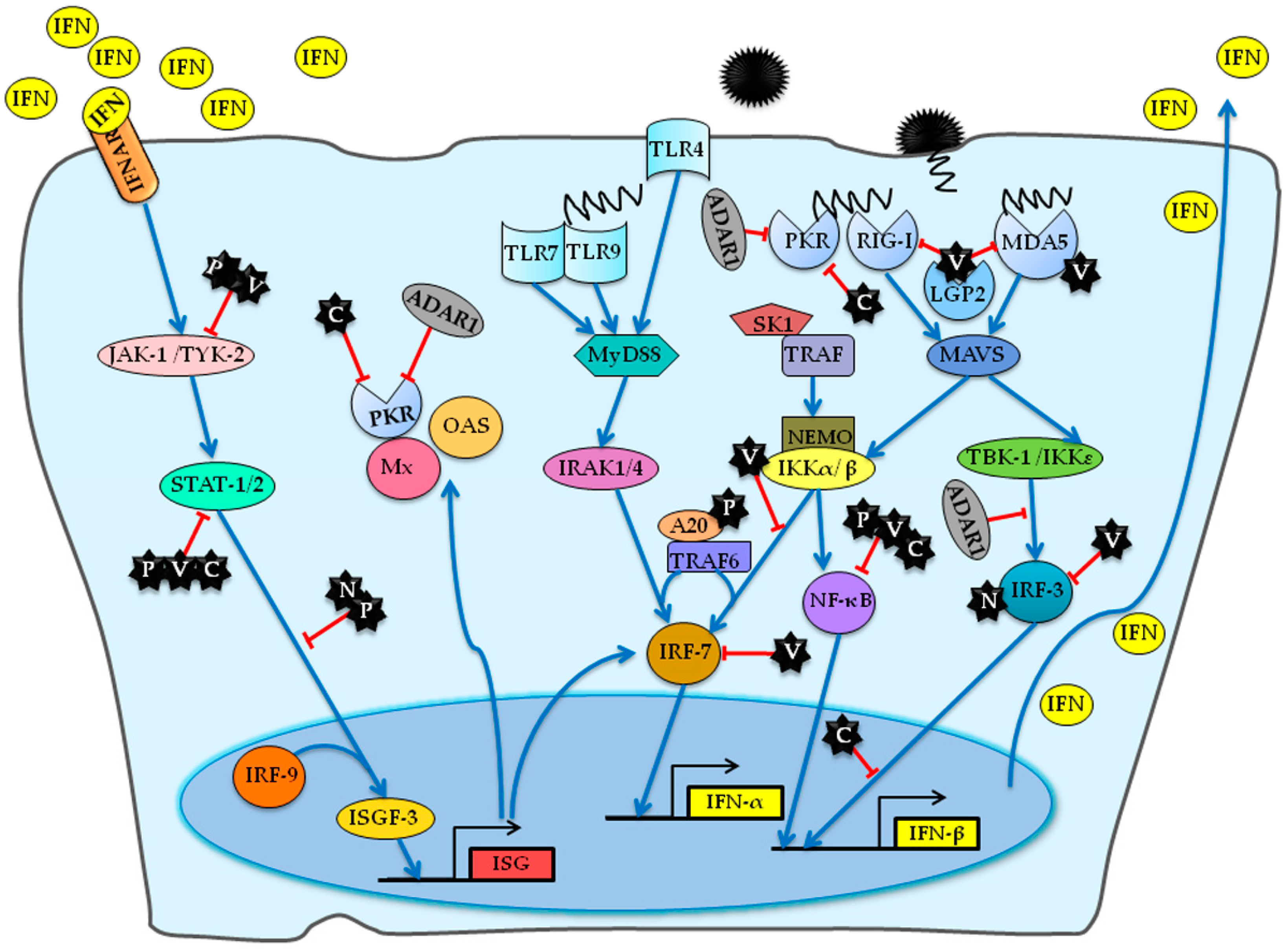
3.2. Host Factors Involved in Anti-MeV Innate Immune Responses
3.2.1. Host Factors Involved in IFN Response
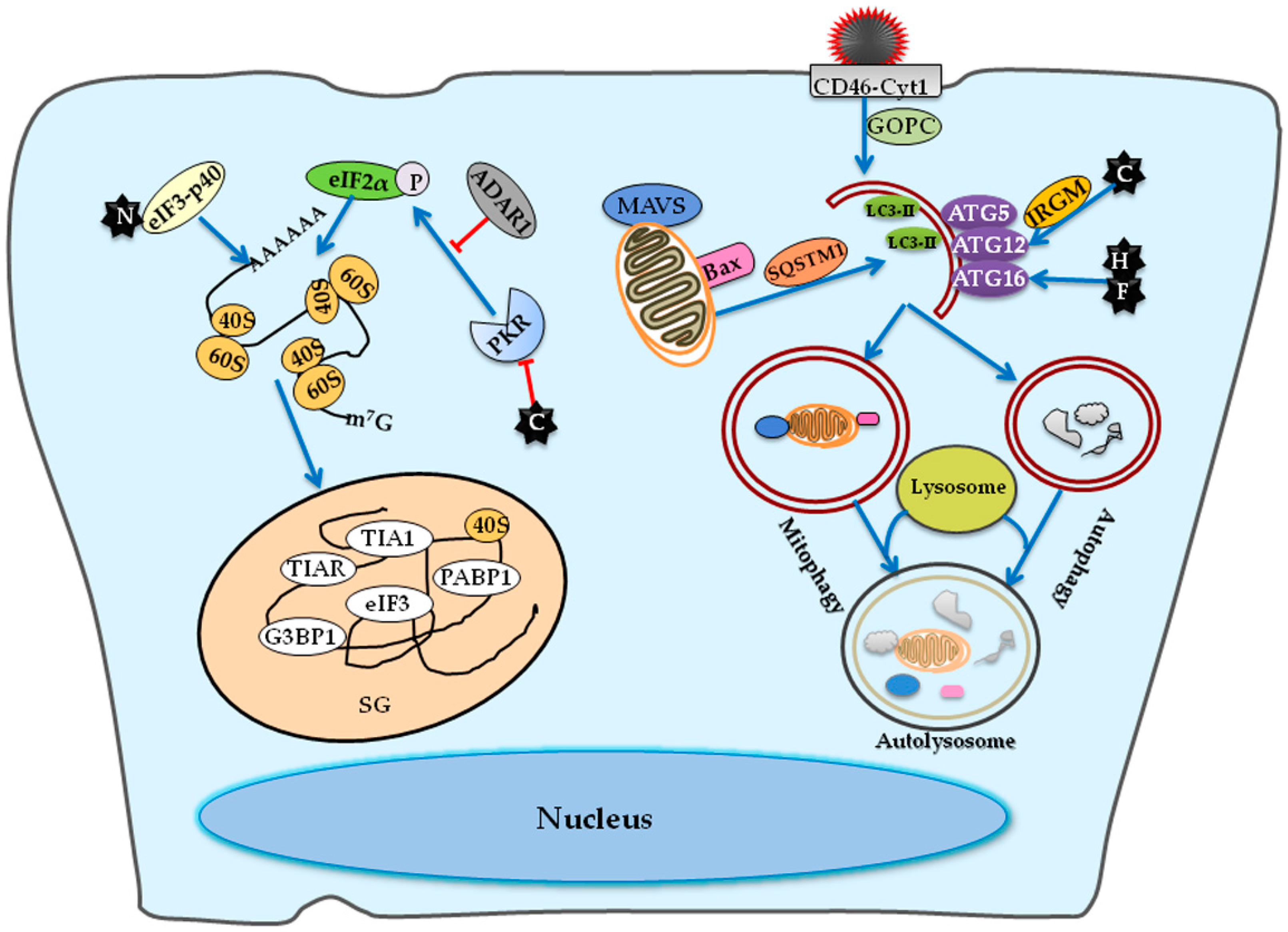
3.2.2. Host Factors Involved in Stress Granule Formation
3.2.3. Host Factors Involved in Autophagy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chesney, R.C.M.M. Measles Virus. In Encyclopedia of Virology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 285–291. [Google Scholar]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Genus Morbillivirus. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier/Academic Press: London, UK, 2011; pp. 680–681. [Google Scholar]
- Goodson, J.L.; Seward, J.F. Measles 50 Years after Use of Measles Vaccine. Infect. Dis. Clin. N. Am. 2015, 29, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, H.; Hengel, H.; Tenbusch, M.; Doerr, H.W. Eradication of measles: Remaining challenges. Med. Microbiol. Immunol. 2016, 205, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Plemper, R.K. The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front. Microbiol. 2015, 6, 459. [Google Scholar] [CrossRef] [PubMed]
- Galinski, M.S. Paramyxoviridae: Transcription and replication. Adv. Virus Res. 1991, 39, 129–162. [Google Scholar] [PubMed]
- tenOever, B.R.; Servant, M.J.; Grandvaux, N.; Lin, R.; Hiscott, J. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 2002, 76, 3659–3669. [Google Scholar] [CrossRef] [PubMed]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles virus fusion orotein: Structure, function and inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E.; Lin, W.H.; Pan, C.H. Measles virus, immune control, and persistence. FEMS Microbiol. Rev. 2012, 36, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Naim, H.Y. Measles virus. Hum. Vaccines Immunother. 2015, 11, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Halsey, N.A. The clinical significance of measles: A review. J. Infect. Dis. 2004, 189 (Suppl. S1), S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Simons, E.; Ferrari, M.; Fricks, J.; Wannemuehler, K.; Anand, A.; Burton, A.; Strebel, P. Assessment of the 2010 global measles mortality reduction goal: Results from a model of surveillance data. Lancet 2012, 379, 2173–2178. [Google Scholar] [CrossRef]
- Van den Ent, M.M.; Brown, D.W.; Hoekstra, E.J.; Christie, A.; Cochi, S.L. Measles mortality reduction contributes substantially to reduction of all cause mortality among children less than five years of age, 1990–2008. J. Infect. Dis. 2011, 204 (Suppl. S1), S18–S23. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.L.; Safdar, N. The reemergence of measles. Curr. Infect. Dis. Rep. 2015, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Caseris, M.; Burdet, C.; Lepeule, R.; Houhou, N.; Yeni, P.; Yazdanpanah, Y.; Joly, V. An update on measles. Rev. Med. Interne 2015, 36, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Brown, K.; Mankertz, A.; Santibanez, S.; Shulga, S.; Muller, C.P.; Hubschen, J.M.; Siqueira, M.; Beirnes, J.; Ahmed, H.; et al. Global distribution of measles genotypes and measles molecular epidemiology. J. Infect. Dis. 2011, 204 (Suppl. S1), S514–S523. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Bellini, W.J. Update on the global distribution of genotypes of wild type measles viruses. J. Infect. Dis. 2003, 187 (Suppl. S1), S270–S276. [Google Scholar] [CrossRef] [PubMed]
- Riddell, M.A.; Rota, J.S.; Rota, P.A. Review of the temporal and geographical distribution of measles virus genotypes in the prevaccine and postvaccine eras. Virol. J. 2005, 2, 87. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Receveur-Brechot, V.; Oglesbee, M.; Zhang, X.; Buccellato, M.; Darbon, H.; Canard, B.; Finet, S.; Longhi, S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005, 14, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Schoehn, G.; Mavrakis, M.; Albertini, A.; Wade, R.; Hoenger, A.; Ruigrok, R.W. The 12 A structure of trypsin-treated measles virus N-RNA. J. Mol. Biol. 2004, 339, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Horikami, S.M.; Moyer, S.A. Structure, transcription, and replication of measles virus. Curr. Top. Microbiol. Immunol. 1995, 191, 35–50. [Google Scholar] [PubMed]
- Dutch, R.E. Paramyxoviruses. In Encyclopedia of Virology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 52–57. [Google Scholar]
- Blocquel, D.; Habchi, J.; Costanzo, S.; Doizy, A.; Oglesbee, M.; Longhi, S. Interaction between the C-terminal domains of measles virus nucleoprotein and phosphoprotein: A tight complex implying one binding site. Protein Sci. 2012, 21, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Masuda, M.; Kanai, M.; Tsukiyama-Kohara, K.; Yoneda, M.; Kai, C. Measles virus N protein inhibits host translation by binding to eIF3-p40. J. Virol. 2007, 81, 11569–11576. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Bourhis, J.M.; Chamontin, C.; Soriano, C.; Villet, S.; Costanzo, S.; Couturier, M.; Belle, V.; Fournel, A.; Darbon, H.; et al. The interaction between the measles virus nucleoprotein and the Interferon Regulator Factor 3 relies on a specific cellular environment. Virol. J. 2009, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Bellini, W.J.; Englund, G.; Rozenblatt, S.; Arnheiter, H.; Richardson, C.D. Measles virus P gene codes for two proteins. J. Virol. 1985, 53, 908–919. [Google Scholar] [PubMed]
- Spehner, D.; Drillien, R.; Howley, P.M. The assembly of the measles virus nucleoprotein into nucleocapsid-like particles is modulated by the phosphoprotein. Virology 1997, 232, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Guryanov, S.G.; Liljeroos, L.; Kasaragod, P.; Kajander, T.; Butcher, S.J. Crystal Structure of the Measles Virus Nucleoprotein Core in Complex with an N-terminal Region of Phosphoprotein. J. Virol. 2016, 90, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Krumm, S.A.; Takeda, M.; Plemper, R.K. The measles virus nucleocapsid protein tail domain is dispensable for viral polymerase recruitment and activity. J. Biol. Chem. 2013, 288, 29943–29953. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.; Chopy, D.; Dosnon, M.; Bloyet, L.M.; Devaux, P.; Urzua, E.; Cattaneo, R.; Longhi, S.; Gerlier, D. Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 2014, 88, 10851–10863. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Canard, B.; Longhi, S. Structural disorder within the replicative complex of measles virus: Functional implications. Virology 2006, 344, 94–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Zhang, G.; Yan, Q.; Yang, X.; Ding, B.; Tang, Q.; Sun, S.; Hu, Z.; Chen, M. An amino acid of human parainfluenza virus type 3 nucleoprotein is critical for template function and cytoplasmic inclusion body formation. J. Virol. 2013, 87, 12457–12470. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Thomas, D.; Lewicki, H.; Billeter, M.A.; Oldstone, M.B. V and C proteins of measles virus function as virulence factors in vivo. Virology 2000, 267, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.; Cattaneo, R. Measles virus phosphoprotein gene products: Conformational flexibility of the P/V protein amino-terminal domain and C protein infectivity factor function. J. Virol. 2004, 78, 11632–11640. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Duprex, W.P.; Krohne, G.; Rima, B.K.; Schneider-Schaulies, S. Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J. Gen. Virol. 2007, 88 Pt 4, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Takeda, M.; Yanagi, Y. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J. Virol. 2007, 81, 6827–6836. [Google Scholar] [CrossRef] [PubMed]
- Reuter, T.; Weissbrich, B.; Schneider-Schaulies, S.; Schneider-Schaulies, J. RNA interference with measles virus N, P, and L mRNAs efficiently prevents and with matrix protein mRNA enhances viral transcription. J. Virol. 2006, 80, 5951–5957. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Banerjee, A.K. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 2007, 25, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Kobayashi, M.; Iwama, M.; Mizumoto, K. Sendai virus RNA-dependent RNA polymerase L protein catalyzes cap methylation of virus-specific mRNA. J. Biol. Chem. 2005, 280, 4429–4435. [Google Scholar] [CrossRef] [PubMed]
- Plumet, S.; Duprex, W.P.; Gerlier, D. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 2005, 79, 6900–6908. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.K.; Duprex, W.P. The measles virus replication cycle. Curr. Top. Microbiol. Immunol. 2009, 329, 77–102. [Google Scholar] [PubMed]
- Brindley, M.A.; Takeda, M.; Plattet, P.; Plemper, R.K. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. USA 2012, 109, E3018–E3027. [Google Scholar] [CrossRef] [PubMed]
- El Najjar, F.; Schmitt, A.P.; Dutch, R.E. Paramyxovirus glycoprotein incorporation, assembly and budding: A three way dance for infectious particle production. Viruses 2014, 6, 3019–3054. [Google Scholar] [CrossRef] [PubMed]
- Colf, L.A.; Juo, Z.S.; Garcia, K.C. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 2007, 14, 1227–1228. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Kajikawa, M.; Maita, N.; Takeda, M.; Kuroki, K.; Sasaki, K.; Kohda, D.; Yanagi, Y.; Maenaka, K. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. USA 2007, 104, 19535–19540. [Google Scholar] [CrossRef] [PubMed]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Runkler, N.; Dietzel, E.; Moll, M.; Klenk, H.D.; Maisner, A. Glycoprotein targeting signals influence the distribution of measles virus envelope proteins and virus spread in lymphocytes. J. Gen. Virol. 2008, 89 Pt 3, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.D.; Leonard, V.H.; Cattaneo, R. The measles virus fusion protein transmembrane region modulates availability of an active glycoprotein complex and fusion efficiency. J. Virol. 2008, 82, 11437–11445. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chan, Y.P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.F.; Skiniotis, G.; et al. Crystal structure of the pre-fusion Nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.P.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar] [PubMed]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.S.; Sakaguchi, T.; Schmitt, A.P. Paramyxovirus assembly and budding: Building particles that transmit infections. Int. J. Biochem. Cell Biol. 2010, 42, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Runkler, N.; Pohl, C.; Schneider-Schaulies, S.; Klenk, H.D.; Maisner, A. Measles virus nucleocapsid transport to the plasma membrane requires stable expression and surface accumulation of the viral matrix protein. Cell. Microbiol. 2007, 9, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhong, Y.; Qin, Y.; Chen, M. Interaction of human parainfluenza virus type 3 nucleoprotein with matrix protein mediates internal viral protein assembly. J. Virol. 2016, 90, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, T.; Portner, A. Molecular mechanism of paramyxovirus budding. Virus Res. 2004, 106, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Liljeroos, L.; Butcher, S.J. Matrix proteins as centralized organizers of negative-sense RNA virions. Front. Biosci. 2013, 18, 696–715. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Salditt, A.; Koethe, S.; Pohl, C.; Harms, H.; Kolesnikova, L.; Becker, S.; Schneider-Schaulies, S. Measles virus M protein-driven particle production does not involve the endosomal sorting complex required for transport (ESCRT) system. J. Gen. Virol. 2010, 91 Pt 6, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.C.; Iorio, C.; Sarangi, F.; Khine, A.A.; Richardson, C.D. CDw150(SLAM) is a receptor for a lymphotropic strain of measles virus and may account for the immunosuppressive properties of this virus. Virology 2001, 279, 9–21. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; McQuaid, S.; van Amerongen, G.; Yuksel, S.; Verburgh, R.J.; Osterhaus, A.D.; Duprex, W.P.; de Swart, R.L. Measles immune suppression: Lessons from the macaque model. PLoS Pathog. 2012, 8, e1002885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyce, R.S.; Richardson, C.D. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol. 2012, 20, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Dorig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Naniche, D.; Varior-Krishnan, G.; Cervoni, F.; Wild, T.F.; Rossi, B.; Rabourdin-Combe, C.; Gerlier, D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993, 67, 6025–6032. [Google Scholar] [PubMed]
- Young, V.A.; Rall, G.F. Making it to the synapse: Measles virus spread in and among neurons. Curr. Top. Microbiol. Immunol. 2009, 330, 3–30. [Google Scholar] [PubMed]
- Laksono, B.M.; de Vries, R.D.; McQuaid, S.; Duprex, W.P.; de Swart, R.L. Measles virus host invasion and pathogenesis. Viruses 2016, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Lyupina, Y.V.; Dmitrieva, S.B.; Timokhova, A.V.; Beljelarskaya, S.N.; Zatsepina, O.G.; Evgen’ev, M.B.; Mikhailov, V.S. An important role of the heat shock response in infected cells for replication of baculoviruses. Virology 2010, 406, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chen, Y.H.; Chow, L.P.; Tsai, Y.H.; Chen, P.H.; Huang, C.Y.; Chen, W.T.; Hwang, L.H. Heat shock protein 72 is associated with the hepatitis C virus replicase complex and enhances viral RNA replication. J. Biol. Chem. 2010, 285, 28183–28190. [Google Scholar] [CrossRef] [PubMed]
- Carsillo, T.; Traylor, Z.; Choi, C.; Niewiesk, S.; Oglesbee, M. Hsp72, a host determinant of measles virus neurovirulence. J. Virol. 2006, 80, 11031–11039. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Iwasaki, M.; Takeda, M.; Nakamura, T.; Yanagi, Y.; Ohno, S. Measles virus nonstructural C protein modulates viral RNA polymerase activity by interacting with host protein SHCBP1. J. Virol. 2013, 87, 9633–9642. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Schuster, A.; Schneider-Schaulies, S.; Banerjee, A.K. Involvement of cellular casein kinase II in the phosphorylation of measles virus P protein: Identification of phosphorylation sites. Virology 1995, 211, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Sugai, A.; Sato, H.; Hagiwara, K.; Kozuka-Hata, H.; Oyama, M.; Yoneda, M.; Kai, C. Newly identified minor phosphorylation site threonine-279 of measles virus nucleoprotein is a prerequisite for nucleocapsid formation. J. Virol. 2014, 88, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Shimodo, M.; Satoh, Y.; Kitagawa, Y.; Takeuchi, K.; Gotoh, B.; Itoh, M. F-actin modulates measles virus cell-cell fusion and assembly by altering the interaction between the matrix protein and the cytoplasmic tail of hemagglutinin. J. Virol. 2013, 87, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Sugai, A.; Sato, H.; Yoneda, M.; Kai, C. Phosphorylation of measles virus phosphoprotein at S86 and/or S151 downregulates viral transcriptional activity. FEBS Lett. 2012, 586, 3900–3907. [Google Scholar] [CrossRef] [PubMed]
- Sugai, A.; Sato, H.; Yoneda, M.; Kai, C. Phosphorylation of measles virus nucleoprotein affects viral growth by changing gene expression and genomic RNA stability. J. Virol. 2013, 87, 11684–11692. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.A.; Baker, S.C.; Lessard, J.L. Tubulin: A factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc. Natl. Acad. Sci. USA 1986, 83, 5405–5409. [Google Scholar] [CrossRef] [PubMed]
- Berghall, H.; Wallen, C.; Hyypia, T.; Vainionpaa, R. Role of cytoskeleton components in measles virus replication. Arch. Virol. 2004, 149, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.A.; Baker, S.C.; Horikami, S.M. Host cell proteins required for measles virus reproduction. J. Gen. Virol. 1990, 71 Pt 4, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Ma, X.; Seki, F.; Suzuki, T.; Iwasaki, M.; Yanagi, Y.; Komase, K.; Takeda, M. Intracellular transport of the measles virus ribonucleoprotein complex is mediated by Rab11A-positive recycling endosomes and drives virus release from the apical membrane of polarized epithelial cells. J. Virol. 2013, 87, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, E.; Kolesnikova, L.; Maisner, A. Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol. J. 2013, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Sánchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; Tangy, F.; et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 2016, 5, e11275. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, S.; Takeda, M.; Ohno, S.; Nakatsu, Y.; Nakanishi, Y.; Yanagi, Y. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 2010, 84, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [PubMed]
- Runge, S.; Sparrer, K.M.; Lassig, C.; Hembach, K.; Baum, A.; Garcia-Sastre, A.; Soding, J.; Conzelmann, K.K.; Hopfner, K.P. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 2014, 10, e1004081. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.R.; Horvath, C.M. Paramyxovirus V protein interaction with the antiviral sensor LGP2 disrupts MDA5 signaling enhancement but is not relevant to LGP2-mediated RLR signaling inhibition. J. Virol. 2014, 88, 8180–8188. [Google Scholar] [CrossRef] [PubMed]
- Uchikawa, E.; Lethier, M.; Malet, H.; Brunel, J.; Gerlier, D.; Cusack, S. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Mol. Cell 2016, 62, 586–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E.; Wang, M.K.; Rennick, L.J.; Full, F.; Gableske, S.; Mesman, A.W.; Gringhuis, S.I.; Geijtenbeek, T.B.; Duprex, W.P.; Gack, M.U. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 2014, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Okabayashi, T.; Yokosawa, N.; Fujii, N. Measles virus P protein suppresses Toll-like receptor signal through up-regulation of ubiquitin-modifying enzyme A20. FASEB J. 2008, 22, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hailey, D.W.; Soetandyo, N.; Li, W.; Lippincott-Schwartz, J.; Shu, H.B.; Ye, Y. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochim. Biophys. Acta 2008, 1783, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Creagh, E.M.; O’Neill, L.A. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Conzelmann, K.K. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J. Virol. 2008, 82, 12365–12373. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Kiyotani, K.; Igarashi, T.; Yoshida, A.; Sakaguchi, T. Inhibition of interferon regulatory factor 3 activation by paramyxovirus V protein. J. Virol. 2012, 86, 7136–7145. [Google Scholar] [CrossRef] [PubMed]
- Komune, N.; Ichinohe, T.; Ito, M.; Yanagi, Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J. Virol. 2011, 85, 13019–13026. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, K.M.; Pfaller, C.K.; Conzelmann, K.K. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 2011, 85, 3162–3171. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Seo, Y.J.; Pritzl, C.J.; Squires, S.A.; Alexander, S.; Hahm, B. Sphingosine kinase 1 regulates measles virus replication. Virology 2014, 450–451, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Radeke, M.J.; Cattaneo, R.; Samuel, C.E. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J. Virol. 2014, 88, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.; Pfaller, C.K.; Conzelmann, K.K. Measles virus C protein interferes with Beta interferon transcription in the nucleus. J. Virol. 2012, 86, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B. Viral Inhibition of the IFN-Induced JAK/STAT Signalling Pathway: Development of Live Attenuated Vaccines by Mutation of Viral-Encoded IFN-Antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Pitini, V.; Arrigo, C.; Altavilla, G. How cells respond to interferons. J. Clin. Oncol. 2010, 28, e439. [Google Scholar] [CrossRef] [PubMed]
- Takayama, I.; Sato, H.; Watanabe, A.; Omi-Furutani, M.; Sugai, A.; Kanki, K.; Yoneda, M.; Kai, C. The nucleocapsid protein of measles virus blocks host interferon response. Virology 2012, 424, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Palosaari, H.; Parisien, J.P.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 2003, 77, 7635–7644. [Google Scholar] [CrossRef] [PubMed]
- Caignard, G.; Bouraï, M.; Jacob, Y.; Tangy, F.; Vidalain, P.O. Inhibition of IFN-α/β signaling by two discrete peptides within measles virus V protein that specifically bind STAT1 and STAT2. Virology 2009, 383, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.; von Messling, V.; Songsungthong, W.; Springfeld, C.; Cattaneo, R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 2007, 360, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.; Hudacek, A.W.; Hodge, G.; Reyes-del Valle, J.; McChesney, M.B.; Cattaneo, R. A recombinant measles virus unable to antagonize STAT1 function cannot control inflammation and is attenuated in Rhesus monkeys. J. Virol. 2011, 85, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.-I.; Okabayashi, T.; Fujii, N. Measles virus C protein suppresses gamma-activated factor formation and virus-induced cell growth arrest. Virology 2011, 414, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okonski, K.M.; Samuel, C.E. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J. Virol. 2012, 86, 3787–3794. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Okonski, K.M.; Samuel, C.E. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 2013, 87, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Takeda, M.; Ohno, S.; Shirogane, Y.; Iwasaki, M.; Yanagi, Y. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 2008, 82, 8296–8306. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Komatsu, T.; Kitagawa, Y.; Sada, K.; Gotoh, B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 2008, 82, 10102–10110. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30 Pt 6, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.X.; Randall, G. Manipulation or capitulation: Virus interactions with autophagy. Microbes Infect. 2012, 14, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Gregoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Delpeut, S.; Rudd, P.A.; Labonte, P.; von Messling, V. Membrane fusion-mediated autophagy induction enhances morbillivirus cell-to-cell spread. J. Virol. 2012, 86, 8527–8535. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Rivailler, P.; Trescol-Biemont, M.C.; Gimenez, C.; Rabourdin-Combe, C.; Horvat, B. Enhanced MHC class II-restricted presentation of measles virus (MV) hemagglutinin in transgenic mice expressing human MV receptor CD46. Eur. J. Immunol. 1998, 28, 1301–1314. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Meng, G.; Jiang, A.; Chen, A.; Dahlhaus, M.; Gonzalez, P.; Beltinger, C.; Wei, J. Mitophagy switches cell death from apoptosis to necrosis in NSCLC cells treated with oncolytic measles virus. Oncotarget 2014, 5, 3907–3918. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Qin, Y.; Chen, M. Host–Pathogen Interactions in Measles Virus Replication and Anti-Viral Immunity. Viruses 2016, 8, 308. https://doi.org/10.3390/v8110308
Jiang Y, Qin Y, Chen M. Host–Pathogen Interactions in Measles Virus Replication and Anti-Viral Immunity. Viruses. 2016; 8(11):308. https://doi.org/10.3390/v8110308
Chicago/Turabian StyleJiang, Yanliang, Yali Qin, and Mingzhou Chen. 2016. "Host–Pathogen Interactions in Measles Virus Replication and Anti-Viral Immunity" Viruses 8, no. 11: 308. https://doi.org/10.3390/v8110308
APA StyleJiang, Y., Qin, Y., & Chen, M. (2016). Host–Pathogen Interactions in Measles Virus Replication and Anti-Viral Immunity. Viruses, 8(11), 308. https://doi.org/10.3390/v8110308