
Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Collection
2.2. RNA Extraction and RT-PCR Amplification
2.3. Cloning and Sequencing
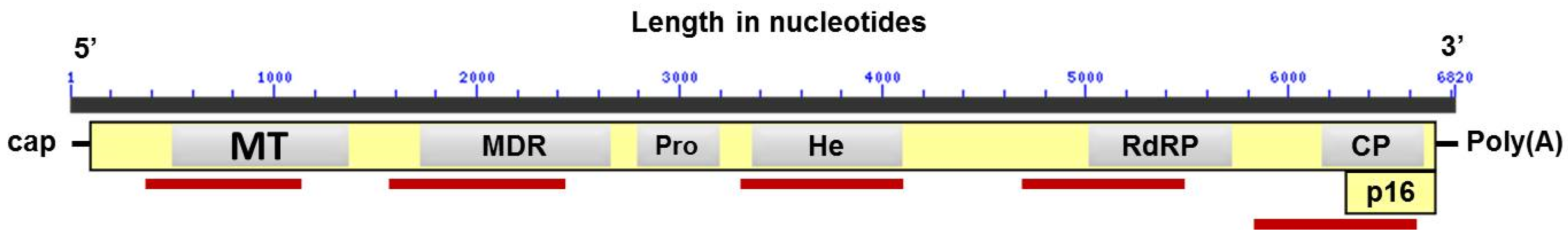
2.4. Nucleotide Sequence Analysis
3. Results
3.1. Genetic Diversity of CSDaV Population
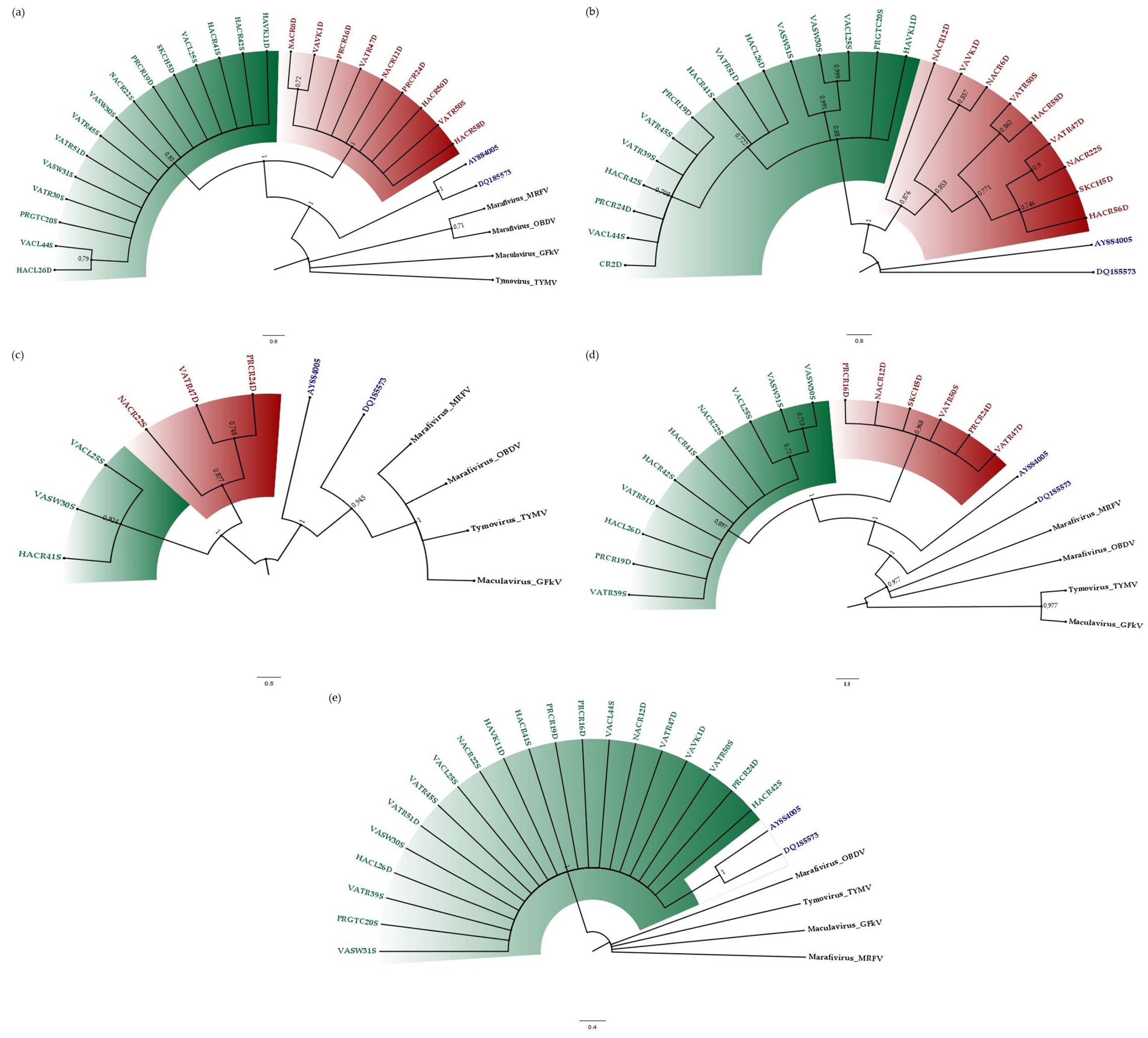
3.2. Phylogenetic Relationships of CSDaV Isolates
3.3. Comparison of Genetic Diversity between Isolates from Asymptomatic and Symptomatic Plants
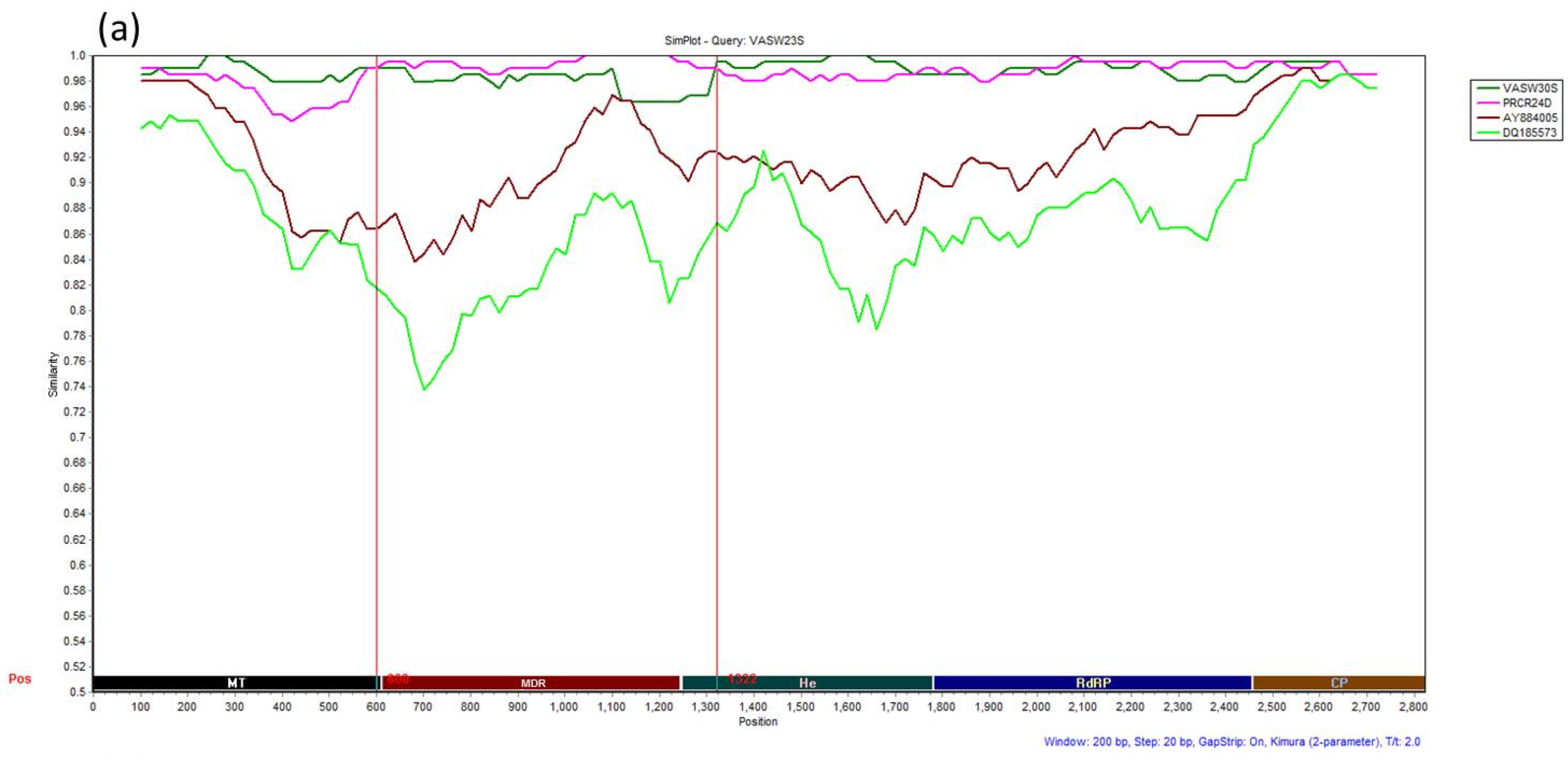
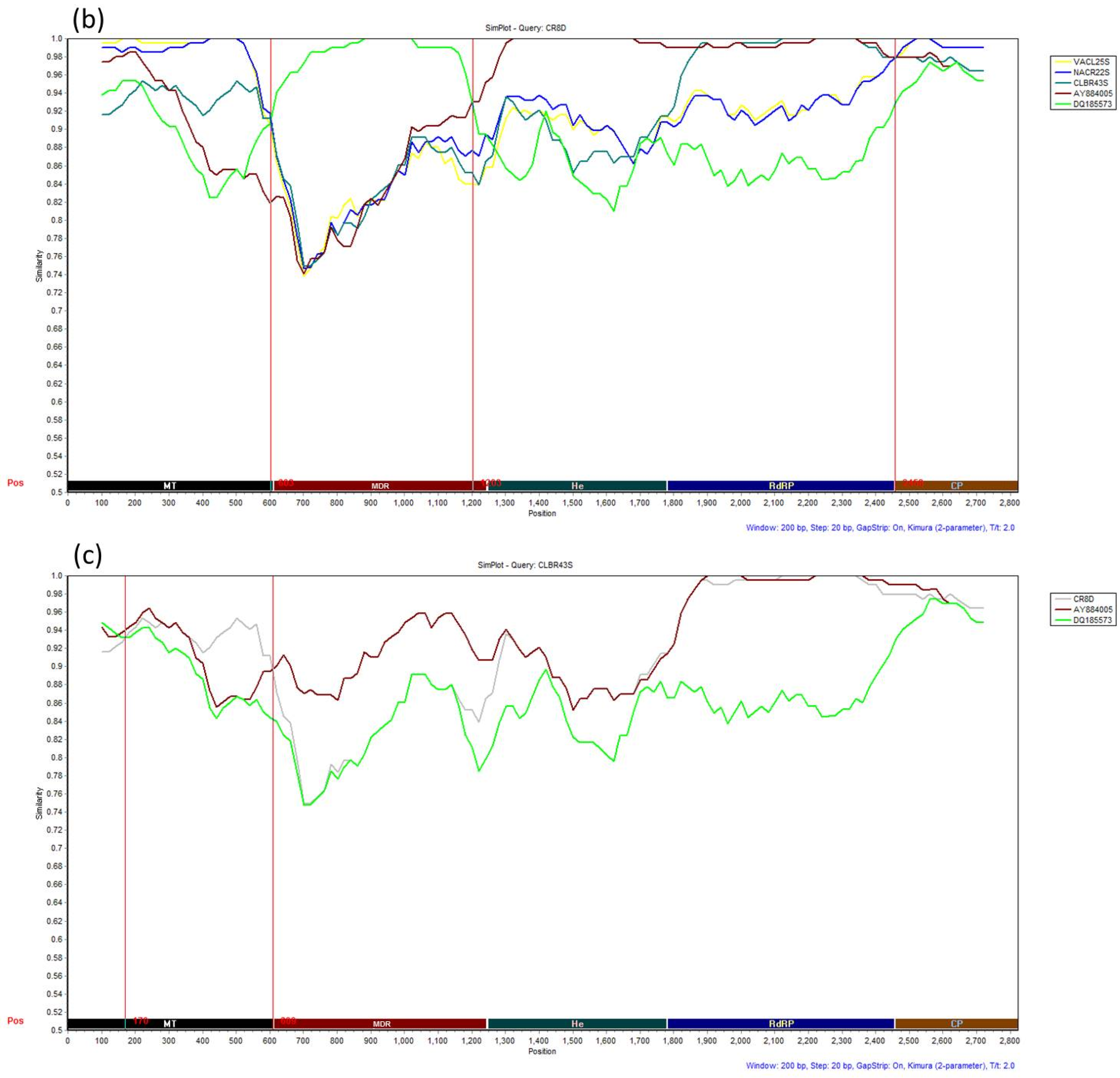
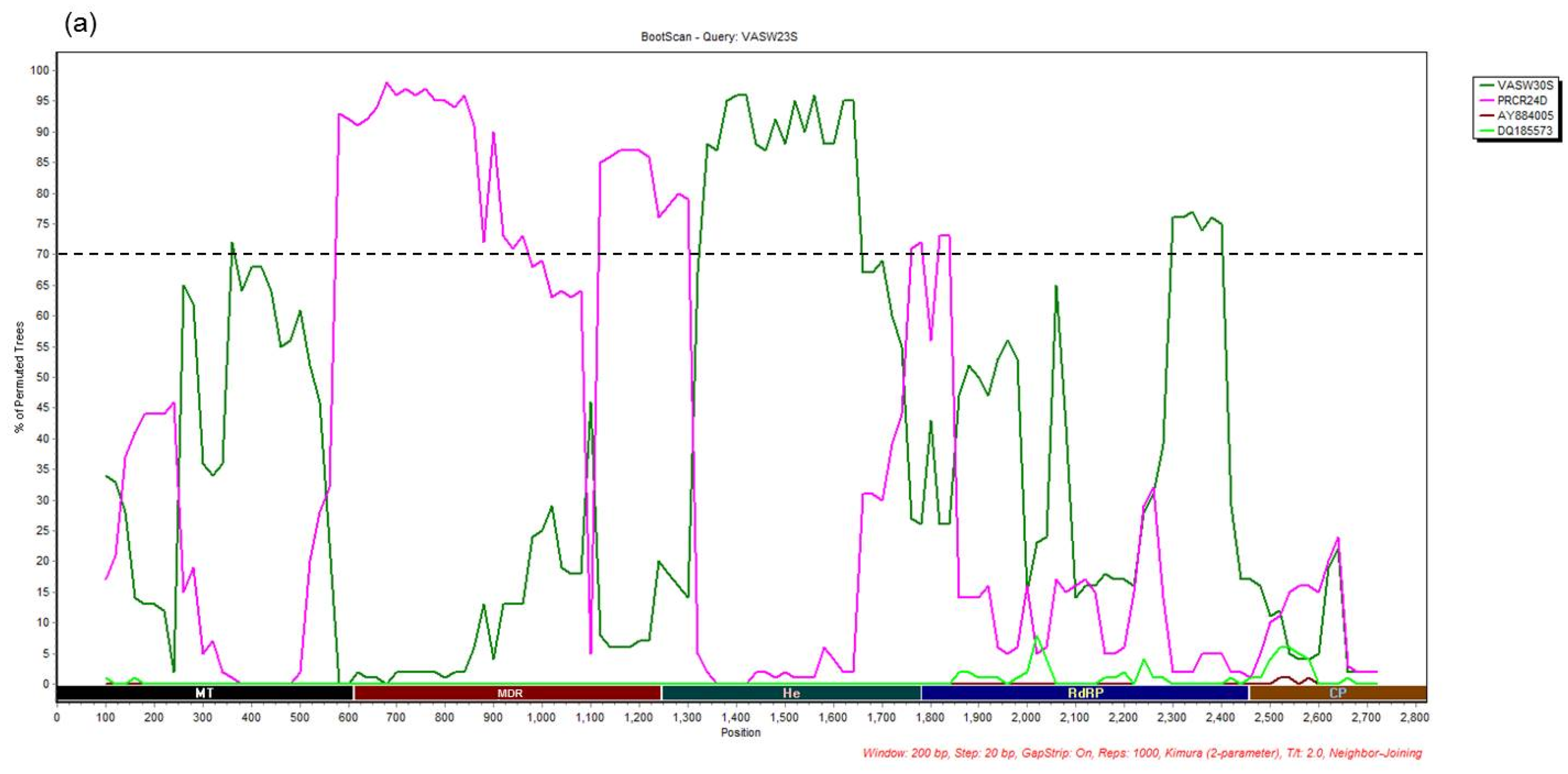
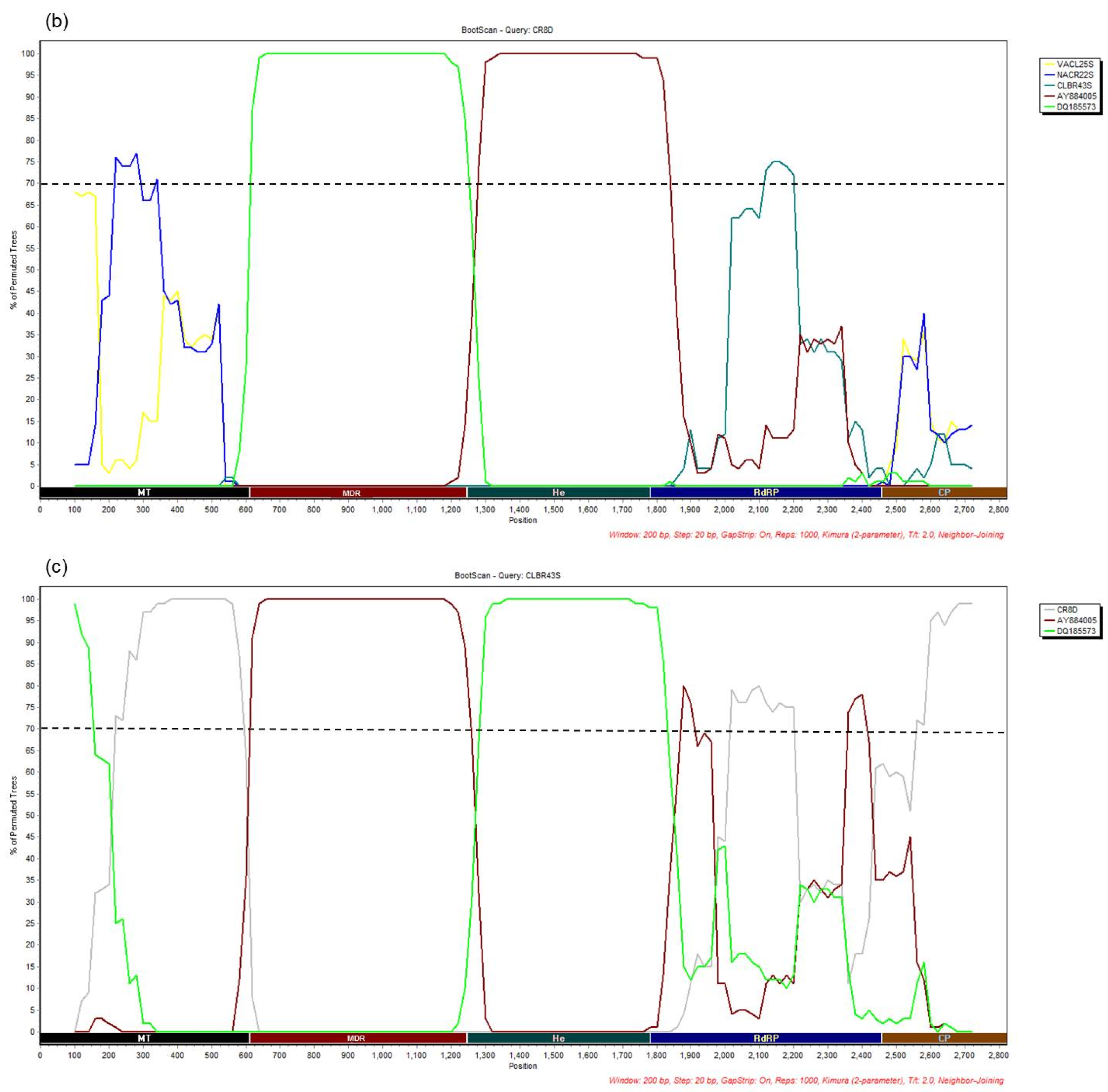
3.4. Recombination Analysis
3.5. Selective Pressure for Different Genomic Regions of CSDaV
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maccheroni, W.; Alegria, M.C.; Greggio, C.C.; Piazza, J.P.; Kamla, R.F.; Zacharias, P.R.A.; Bar-Joseph, M.; Kitajima, E.W.; Assumpção, L.C.; Camarotte, G.; et al. Identification and genomic characterization of a new virus (Tymoviridae family) associated with citrus sudden death disease. J. Virol. 2005, 79, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Mahy, B.W.J.; van Regenmortel, M.H.V. Emerging and reemerging virus diseases of plants. In Desk Encyclopedia of Plant and Fungal Virology, 1st ed.; Mahy, B.W.J., van Regenmortel, M.H.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 412–417. [Google Scholar]
- Müller, G.W.; de Negri, J.D.; Aguilar-Vildoso, C.I.; Mattos, D., Jr.; Pompeu, J., Jr.; Teófilo Sobrinho, J.; Machado, M.A. Citrus sudden death: A new citrus disease in Brazil. In XV Conference of the International Organization of Citrus Virologists, Paphos, Chipre, 2001; p. 100.
- Gomes, C.P.C.; Nagata, T.; Jesus Junior, W.C.; Borges Neto, C.R.; Pappas Junior, G.J.; Martin, D.P. Genetic variation and recombination of RdRp and HSP 70h genes of Citrus tristeza virus isolates from orange trees showing symptoms of citrus sudden death disease. Virol. J. 2008, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta Filho, H.D.; Centro de Citricultura Sylvio Moreira, Instituto Agronômico, Cordeiropolis, SP, Brazil. Personal communication, 2014.
- Bassanezi, R.B.; Montesino, L.H.; Sanches, A.L.; Spósito, M.B.; Stuchi, E.S.; Barbosa, J.C. Effect of citrus sudden death on yield and quality of sweet orange cultivars in Brazil. Plant Dis. 2007, 91, 1407–1412. [Google Scholar] [CrossRef]
- Pantaleo, V.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using high throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar] [PubMed]
- Villamor, D.E.V.; Mekuria, T.A.; Pillai, S.S.; Eastwell, K.C. High throughput sequencing identifies novel viruses in nectarine: Insights to the etiology of stem pitting disease. Phytopathology 2016, 106, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Román, M.P.; Cambra, M.; Juarez, J.; Moreno, P.; Duran-Vila, N.; Tanaka, F.A.O.; Alves, E.; Kitajima, E.W.; Yamamoto, P.T.; Bassanezi, R.B.; et al. Sudden death of citrus in Brazil: A graft-transmissible bud union disease. Plant Dis. 2004, 88, 453–467. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- NCBI: National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 18 August 2016).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; Mcwilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 18 August 2016).
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Lole, S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Fulllength human immunodeficiencey virus Type 1 genomes from subtype Cinfected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Datamonkey: Rapid Detection of Positive Selection. Available online: http://www.datamonkey.org/dataupload.php (accessed on 18 August 2016).
- Nouri, S.; Arevalo, R.; Falk, B.W.; Groves, R.L. Genetic structure and molecular variability of cucumber mosaic virus isolates in the United States. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, E.E.; Centro de Citricultura Sylvio Moreira, Instituto Agronômico, Cordeirópolis, SP, Brazil. Electrophoresis agarose gel showing the size of the amplicons obtained from the PCR of the regions MT, MDR, He, RdRP and CP. 2016. [Google Scholar]
- Matsumura, E.E.; Coletta-Filho, H.D.; et al. Deep sequencing analysis of RNAs from citrus plants grown in citrus sudden death-affected regions reveals diverse known and putative novel viruses. Virology, submitted.
- Garcia-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol 2001, 39, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Barros, C.C.P. Sequenciamento do Genoma Completo e Expressão Heteróloga da Capa Protéica do Marafivirus Associado a Morte Súbita Dos Citros. Ph.D. Thesis, Universidade Católica de Brasilia, Brasília, Brazil, 2006. [Google Scholar]
- Kong, P.; Rubio, L.; Polek, M.; Falk, W.B. Population structure and genetic diversity within California Citrus tristeza virus (CTV) isolates. Virus Genes 2000, 21, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Ayllón, M.A.; Kong, P.; Fernández, A.; Polek, M.; Guerri, J.; Moreno, P.; Falk, B.W. Genetic variation of Citrus tristeza virus isolates from California and Spain: Evidence for mixed infections and recombination. J. Virol. 2001, 75, 8054–8062. [Google Scholar] [CrossRef] [PubMed]
- Ale-Agha, G.N.; Rakhshandehroo, F. Detection and molecular variability of fig fleck-associated virus and fig cryptic virus in Iran. J. Phytopathol. 2013, 162, 417–425. [Google Scholar] [CrossRef]
- Ogawa, T.; Tomitaka, Y.; Nakagawa, A.; Ohshimab, K. Genetic structure of a population of Potato virus Y inducing potato tuber necrotic ringspot disease in Japan; comparison with North American and European populations. Virus Res. 2008, 131, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.K.; Ohshima, K.; Lee, H.G.; Son, M.; Choi, H.S.; Lee, S.H.; Sohn, S.H.; Kim, K.H. Molecular variability and genetic structure of the population of Soybean mosaic virus based on the analysis of complete genome sequences. Virology 2009, 393, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.M.; Malpica, J.M.; Dı́az-Pendon, J.A.; Moriones, E.; Fraile, A.; Garcia-Arenala, F. Variability and genetic structure of the population of watermelon mosaic virus infecting melon in Spain. Virology 2004, 318, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Walia, J.J.; Willemsen, A.; Elci, E.; Caglayan, K.; Falk, B.F.; Rubio, L. Genetic variation and possible mechanisms driving the evolution of worldwide Fig mosaic virus isolates. Phytopathology 2013, 104, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Roossinck, M.J. Genetic bottlenecks. In Plant Virus Evolution; Roossinck, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 123–131. [Google Scholar]
- Li, H.; Roossinck, M.J. Genetic bottlenecks reduce population variation in an experimental RNA virus population. J. Virol. 2004, 78, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Hammond, R.W.; Kogel, R.; Ramirez, P. Variability of geographically distinct isolates of maize rayado fino virus in Latin America. J. Gen. Virol. 1997, 78, 3153–3159. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; den Boon, J.A.; Ahlquist, P. Helicase and capping enzyme active site mutations in brome mosaic virus protein 1a cause defects in template recruitment, negative-strand RNA synthesis, and viral RNA capping. J. Virol. 2000, 74, 8803–8811. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Cameron, C.E. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 2008, 320, 137–156. [Google Scholar]
- Moury, B. Differential selection of genes of cucumber mosaic virus subgroups. Mol. Biol. Evol. 2004, 21, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Jridi, C.; Martin, J.F.; Mareie-Jeanne, V.; Labonne, G.; Blanc, S. Distinct viral populations differentiate and evolve independently in a single perennial host plant. J. Virol. 2006, 80, 2349–2357. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Canopy (C. sinensis) | Rootstock | Collected Tissue | Number of Plants | |
|---|---|---|---|---|
| Asymptomatic plants | Natal | Rangpur lime (C. limonia) | Leaves | 1 |
| Valencia | Swingle citrumelo (P. trifoliate x C. paradisi) | Leaves | 3 | |
| Hamlin | Rangpur lime (C. limonia) | Leaves | 3 | |
| Pera Rio | Gou Tou (unidentified Citrus hybrid) | Leaves | 1 | |
| Valencia | Cleopatra mandarin (C. reshni) | Leaves | 2 | |
| Valencia | Trifoliate orange (P. trifoliata) | Leaves | 3 | |
| Hamlin | Cleopatra mandarin (C. reshni) | Leaves | 1 | |
| Hamlin | Cleopatra mandarin (C. reshni) | Roots | 1 | |
| Symptomatic plants | Valencia | Volkamerian lemon (C. volkameriana) | Leaves | 1 |
| Natal | Rangpur lime (C. limonia) | Leaves | 2 | |
| Hamlin | Rangpur lime (C. limonia) | Leaves | 2 | |
| Hamlin | Volkamerian lemon (C. volkameriana) | Leaves | 1 | |
| Valencia | Rangpur lime (C. limonia) and Trifoliate orange (P. trifoliata) as interstock | Leaves | 2 | |
| Pera Rio | Rangpur lime (C. limonia) | Leaves | 3 | |
| Hamlin | Rangpur lime (C. limonia) and Cleopatra mandarin (C. reshni) as interstock | Leaves | 2 | |
| Hamlin | Rangpur lime (C. limonia) | Roots | 2 | |
| Valencia | Sunki mandarin of China (C. sunki) | Leaves | 1 | |
| Total = 31 plants | ||||
| Genomic Region | Primer Sequences (5’–3’) | Annealing Nucleotide Position |
|---|---|---|
| MT | Forward-CGTCAAACTCCCNCTGAC | 351–368 |
| Reverse-GATCANNAGAGAGTGGACTG | 1094–1113 | |
| MDR | Forward-CTCCCTCTCCATCTGCAAGC | 1566–1585 |
| Reverse-ATANTCNNTGGAGGGGTCA | 2375–2393 | |
| He | Forward-AGATNTTGGCNCTNGANTC | 3305–3323 |
| Reverse-ANTCNGAGAACATTCNGTTG | 4092–4111 | |
| RdRP | Forward-CATCAAGAGAANCANGANCC | 4636–4355 |
| Reverse-TGAGACCATAGTGGGAGTGT | 5414–5433 | |
| CP | Forward-GCCATCTACACCACACTCTC | 5857–5876 |
| Reverse-TTGGANTAGACGGAGTAGGA | 6568–6587 |
| Isolate Identification * | Viral Genomic Region | GenBank Accession No. |
|---|---|---|
| VAVK1D | MT | KX753236 |
| MDR | KX753259 | |
| CP | KX753328 | |
| CR2D | MDR | KX753263 |
| HAVK11D | MT | KX753252 |
| MDR | KX753260 | |
| CP | KX753326 | |
| NACR12D | MT | KX753233 |
| MDR | KX753261 | |
| RdRP | KX753309 | |
| CP | KX753327 | |
| PRCR19D | MT | KX753245 |
| MDR | KX753262 | |
| RdRP | KX753306 | |
| CP | KX753330 | |
| PRGTC20S | MT | KX753254 |
| MDR | KX753264 | |
| CP | KX753321 | |
| VASW23S | MT | KX753248 |
| MDR | KX753265 | |
| He | KX753296 | |
| RdRP | KX753316 | |
| CP | KX753340 | |
| PRCR24D | MT | KX753234 |
| MDR | KX753266 | |
| He | KX753292 | |
| RdRP | KX753313 | |
| CP | KX753336 | |
| HACL26D | MT | KX753243 |
| MDR | KX753267 | |
| RdRP | KX753307 | |
| CP | KX753323 | |
| CR8D 1 | MT | KX753257 |
| MDR | KX753268 | |
| He | KX753297 | |
| RdRP | KX753318 | |
| CP | KX753342 | |
| VASW30S | MT | KX753256 |
| MDR | KX753269 | |
| He | KX753293 | |
| RdRP | KX753299 | |
| CP | KX753324 | |
| VASW31S | MT | KX753242 |
| MDR | KX753270 | |
| RdRP | KX753298 | |
| CP | KX753319 | |
| HACL38S | MT | KX753244 |
| MDR | KX753271 | |
| RdRP | KX753301 | |
| CP | KX753320 | |
| VATR39S | MT | KX753255 |
| MDR | KX753272 | |
| RdRP | KX753305 | |
| CP | KX753322 | |
| HACR42S | MT | KX753241 |
| MDR | KX753273 | |
| RdRP | KX753302 | |
| CP | KX753333 | |
| CLBR43S 2 | MT | KX753251 |
| MDR | KX753274 | |
| He | KX753294 | |
| RdRP | KX753317 | |
| CP | KX753334 | |
| VACL44S | MT | KX753247 |
| MDR | KX753275 | |
| CP | KX753341 | |
| VATR45S | MT | KX753239 |
| MDR | KX753276 | |
| CP | KX753332 | |
| VATR47D | MT | KX753253 |
| MDR | KX753277 | |
| He | KX753291 | |
| RdRP | KX753314 | |
| CP | KX753343 | |
| SKCH5D 3 | MT | KX753237 |
| MDR | KX753278 | |
| RdRP | KX753310 | |
| VATR50S | MT | KX753249 |
| MDR | KX753279 | |
| RdRP | KX753312 | |
| CP | KX753329 | |
| VATR51D | MT | KX753258 |
| MDR | KX753280 | |
| RdRP | KX753304 | |
| CP | KX753325 | |
| HACL52D | MDR | KX753281 |
| RdRP | KX753315 | |
| HACR55S | MDR | KX753282 |
| CP | KX753331 | |
| HACR56D | MT | KX753235 |
| MDR | KX753283 | |
| HACR58D | MT | KX753250 |
| MDR | KX753284 | |
| NACR6D | MT | KX753232 |
| MDR | KX753285 | |
| VACL25S | MT | KX753238 |
| MDR | KX753286 | |
| He | KX753289 | |
| RdRP | KX753300 | |
| CP | KX753337 | |
| NACR22S | MT | KX753246 |
| MDR | KX753287 | |
| He | KX753295 | |
| RdRP | KX753308 | |
| CP | KX753339 | |
| HACR41S | MT | KX753240 |
| MDR | KX753288 | |
| He | KX753290 | |
| RdRP | KX753303 | |
| CP | KX753338 | |
| PRCR16D | MT | KX753231 |
| RdRP | KX753311 | |
| CP | KX753335 |
| Genomic Regions | Number of Final Sequences | S | η | π | θw | dN | dS | ω (dN/dS) |
|---|---|---|---|---|---|---|---|---|
| MT | 28 | 82 | 84 | 0.01815 | 0.0346 | 0.005 ± 0.002 | 0.054 ± 0.010 | 0.093 |
| MDR | 30 | 180 | 214 | 0.04091 | 0.07212 | 0.023 ± 0.004 | 0.097 ± 0.012 | 0.237 |
| He | 9 | 81 | 83 | 0.04185 | 0.05613 | 0.006 ± 0.002 | 0.153 ± 0.020 | 0.039 |
| RdRP | 21 | 70 | 72 | 0.01955 | 0.02895 | 0.001 ± 0.001 | 0.068 ± 0.009 | 0.015 |
| CP | 25 | 26 | 27 | 0.01013 | 0.01897 | 0.003 ± 0.001 | 0.026 ± 0.007 | 0.115 |
| Number of Isolates from Symptomatic Plants/Number of Isolates from Asymptomatic Plants | ||
|---|---|---|
| Group I * | Group II ** | |
| MT | 5/10 | 8/1 |
| MDR | 6/9 | 7/2 |
| He | 0/3 | 2/1 |
| RdRP | 3/7 | 5/1 |
| Symptoms | Number of Sequences | π | θw | dN | dS | ω | |
|---|---|---|---|---|---|---|---|
| MT | Symp. | 13 | 0.01726 | 0.02117 | 0.007 ± 0.002 | 0.044 ± 0.011 | 0.159091 |
| Asymp. | 11 | 0.00770 | 0.01402 | 0.002 ± 0.001 | 0.020 ± 0.005 | 0.100000 | |
| MDR | Symp. | 13 | 0.03441 | 0.04143 | 0.026 ± 0.005 | 0.057 ± 0.011 | 0.456140 |
| Asymp. | 11 | 0.02268 | 0.02601 | 0.014 ± 0.004 | 0.042 ± 0.009 | 0.333333 | |
| He | Symp. | 2 | 0.00942 | 0.00942 | 0.002 ± 0.002 | 0.023 ± 0.012 | 0.086957 |
| Asymp. | 4 | 0.00879 | 0.00924 | 0.003 ± 0.002 | 0.023 ± 0.008 | 0.130435 | |
| RdRP | Symp. | 8 | 0.00856 | 0.00861 | 0.001 ± 0.001 | 0.026 ± 0.008 | 0.038462 |
| Asymp. | 8 | 0.00787 | 0.00918 | 0.001 ± 0.001 | 0.024 ± 0.007 | 0.041667 | |
| CP | Symp. | 9 | 0.00781 | 0.00912 | 0.003 ± 0.001 | 0.019 ± 0.008 | 0.157895 |
| Asymp. | 11 | 0.00862 | 0.01317 | 0.004 ± 0.002 | 0.018 ± 0.006 | 0.222222 |
| Domain | Number of Amino Acid Changes | Total Number of Amino Acid | Position of Amino Acid Changes (Asymp→Symp) |
|---|---|---|---|
| MT | 8 | 202 | 13 (I→T); 57 (P→Q); 113 (Q→R); 144 (L→V); 152 (S→*); 157 (R → K); 171 (A→V) and 199 (T→I) |
| MDR | 10 | 209 | 13 (G→D); 14 (P→R); 22 (L→A); 29 (I→T); 103 (F→S); 109 (F→S); 110 (Q→P; 187 (S→L); 197 (H→R) and 209 (Q→R) |
| He | 4 | 176 | 28 (V→A); 62 (L→P); 72 (T→I) and 120 (M→I) |
| RdRP | 4 | 223 | 37 (P→L); 136 (A→V); 155 (N→ S) and 200 (L→P) |
| CP | 1 | 120 | 55 (Q→R) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumura, E.E.; Coletta Filho, H.D.; De Oliveira Dorta, S.; Nouri, S.; Machado, M.A. Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil. Viruses 2016, 8, 330. https://doi.org/10.3390/v8120330
Matsumura EE, Coletta Filho HD, De Oliveira Dorta S, Nouri S, Machado MA. Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil. Viruses. 2016; 8(12):330. https://doi.org/10.3390/v8120330
Chicago/Turabian StyleMatsumura, Emilyn Emy, Helvécio Della Coletta Filho, Silvia De Oliveira Dorta, Shahideh Nouri, and Marcos Antonio Machado. 2016. "Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil" Viruses 8, no. 12: 330. https://doi.org/10.3390/v8120330
APA StyleMatsumura, E. E., Coletta Filho, H. D., De Oliveira Dorta, S., Nouri, S., & Machado, M. A. (2016). Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil. Viruses, 8(12), 330. https://doi.org/10.3390/v8120330