
Human Cytomegalovirus Strategies to Maintain and Promote mRNA Translation

{kind=link}
Abstract
:1. Introduction
2. The Scanning Model of Translation Initiation
3. Alternative Translation Initiation Mechanisms
4. Signaling Pathways Regulating Translation Initiation
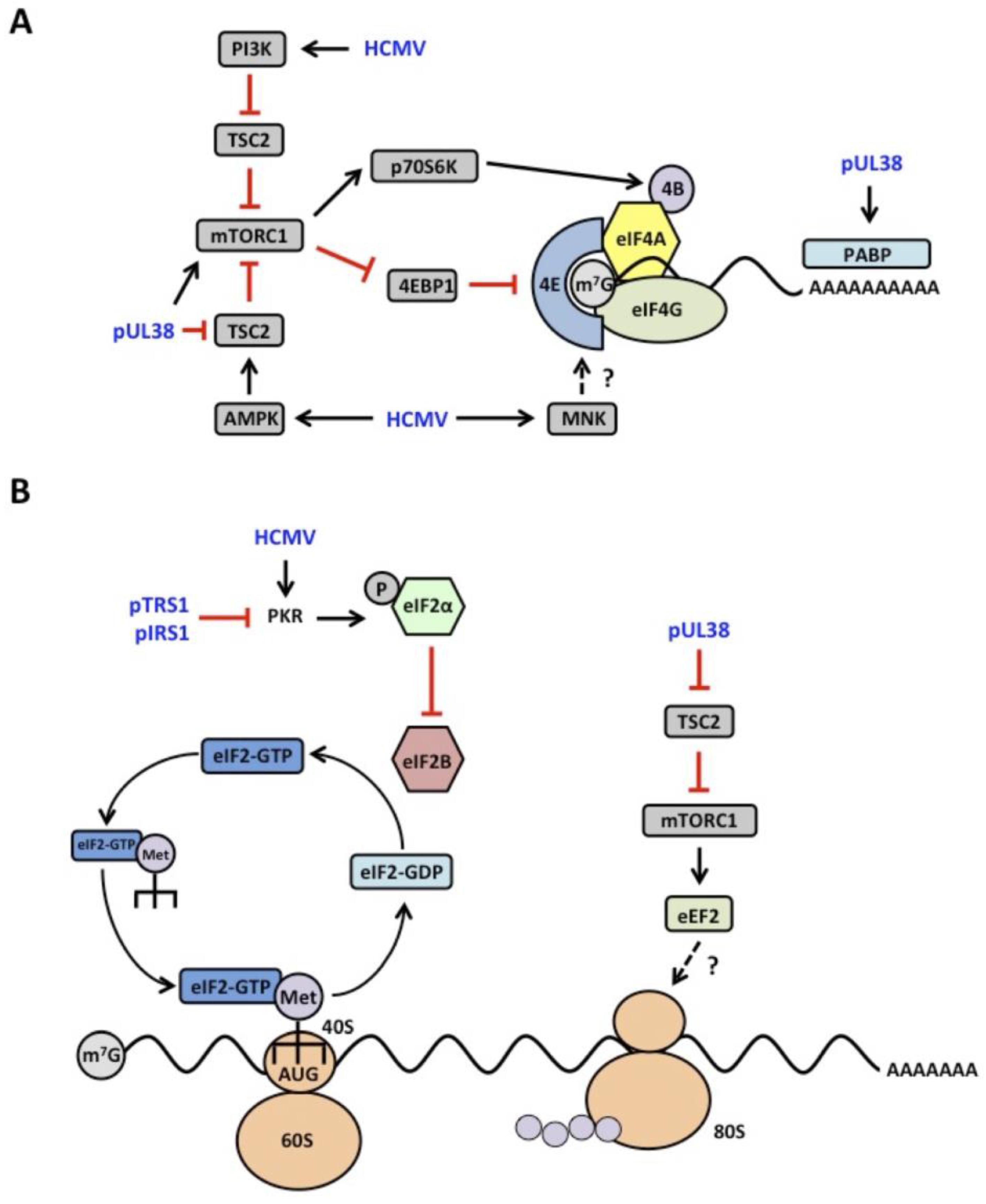
4.1. mTOR Signaling
4.2. eIF2α Kinase Activation
5. HCMV Manipulation of Translation Initiation
5.1 HCMV Infection Increases eIF4F Abundance and Activity during Infection
5.2. IRES Activity during HCMV Infection
5.3. HCMV Regulation of eIF2α Kinases
6. Unresolved Questions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HCMV | human cytomegalovirus |
| eIF4F | eukaryotic initiation factor 4F |
| m7G cap | 7-methyl guanosine cap |
| 43S PIC | 43S preinitiation complex |
| UTR | untranslated region |
| eEF1 | eukaryotic elongation factor 1 |
| eEF2 | eukaryotic elongation factor 2 |
| eIF4E | eukaryotic initiation factor 4E |
| eIF4G | eukaryotic initiation factor 4G |
| eIF4A | eukaryotic initiation factor 4A |
| PABP | poly(A) binding protein |
| eIF1 | eukaryotic initiation factor 1 |
| eIF1A | eukaryotic initiation factor 1A |
| eIF3 | eukaryotic initiation factor 3 |
| eIF4B | eukaryotic initiation factor 4B |
| eIF5 | eukaryotic initiation factor 5 |
| eIF2 | eukaryotic initiation factor 2 |
| tRNAMet | methionyl-tRNA |
| eRF-1 | eukaryotic release factor 1 |
| eRF-3 | eukaryotic release factor 3 |
| IRES | Internal Ribosome Entry Sites |
| ITAF | IRES trans-activating factor |
| PTB | polypyrimidine tract binding protein |
| La | lupus autoantigen |
| HIV | human immunodeficiency virus |
| HCV | hepatitis C virus |
| mTOR | mammalian target of rapamycin kinase |
| 4EBP1 | eIF4E-binding protein 1 |
| p70S6K | 70 kDa ribosomal protein S6 kinase |
| eEF2K | eukaryotic elongation factor 2 kinase |
| TSC | tuberous sclerosis complex |
| TSC1 | tuberous sclerosis complex 1 |
| TSC2 | tuberous sclerosis complex 2 |
| GAP | GTPase activating protein |
| AMPK | AMP-regulated protein kinase |
| PI3K | phosphoinositide 3-kinase |
| AKT | protein kinase B |
| eIF2α | eukaryotic initiation factor 2 alpha subunit |
| eIF2B | eukaryotic initiation factor 2B |
| HRI | heme-regulated inhibitor |
| ER | endoplasmic reticulum |
| PERK | PKR-like endoplasmic reticulum kinase |
| GCN2 | general control nonderepressible 2 |
| PKR | protein kinase R |
| dsRNA | double stranded RNA |
| UPR | unfolded protein response |
| BiP | binding immunoglobulin protein |
| ORF | open reading frame |
| uORF | upstream open reading frame |
| TSS | transcription start site |
References
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.M.; Morgan, M.A.; Shatkin, A.J. Two forms of purified m7G-cap binding protein with different effects on capped mRNA translation in extracts of uninfected and poliovirus-infected HeLa cells. J. Biol. Chem. 1981, 256, 7691–7694. [Google Scholar] [PubMed]
- Grifo, J.A.; Tahara, S.M.; Morgan, M.A.; Shatkin, A.J.; Merrick, W.C. New initiation factor activity required for globin mRNA translation. J. Biol. Chem. 1983, 258, 5804–5810. [Google Scholar] [PubMed]
- Sonenberg, N.; Shatkin, A.J. Nonspecific effect of m7GMP on protein-RNA interactions. J. Biol. Chem. 1978, 253, 6630–6632. [Google Scholar] [PubMed]
- Thomas, A.; Spaan, W.; van Steeg, H.; Voorma, H.O.; Benne, R. Mode of action of protein synthesis initiation factor eIF-1 from rabbit reticulocytes. FEBS Lett. 1980, 116, 67–71. [Google Scholar] [CrossRef]
- Carlberg, U.; Nilsson, A.; Nygard, O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990, 191, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.G.; Selmer, M.; Dunham, C.M.; Weixlbaumer, A.; Kelley, A.C.; Ramakrishnan, V. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science 2009, 326, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Kimata, Y.; Kohno, K. Elongation factor 2 mutants deficient in diphthamide formation show temperature-sensitive cell growth. J. Biol. Chem. 1994, 269, 13497–13501. [Google Scholar] [PubMed]
- Stansfield, I.; Kushnirov, V.V.; Jones, K.M.; Tuite, M.F. A conditional-lethal translation termination defect in a sup45 mutant of the yeast Saccharomyces cerevisiae. Eur. J. Biochem. 1997, 245, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [PubMed]
- De Benedetti, A.; Rhoads, R.E. Overexpression of eukaryotic protein synthesis initiation factor 4E in HeLa cells results in aberrant growth and morphology. Proc. Natl. Acad. Sci. USA 1990, 87, 8212–8216. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Penman, S. Regulation of protein synthesis in mammalian cells. J. Mol. Biol. 1970, 50, 655–670. [Google Scholar] [CrossRef]
- Singer, R.H.; Penman, S. Biological sciences: Stability of HeLa cell mRNA in actinomycin. Nature 1972, 240, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Imataka, H.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Mol. Cell. Biol. 1997, 17, 6940–6947. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Trachsel, H.; Hecht, S.; Shatkin, A.J. Differential stimulation of capped mRNA translation in vitro by cap binding protein. Nature 1980, 285, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Marcotrigiano, J.; Gingras, A.C.; Sonenberg, N.; Burley, S.K. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eiIF4G. Mol. Cell 1999, 3, 707–716. [Google Scholar] [CrossRef]
- Slepenkov, S.V.; Korneeva, N.L.; Rhoads, R.E. Kinetic mechanism for assembly of the m7GPPPG.eIF4E.eIF4GE complex. J. Biol. Chem. 2008, 283, 25227–25237. [Google Scholar] [CrossRef] [PubMed]
- Hinton, T.M.; Coldwell, M.J.; Carpenter, G.A.; Morley, S.J.; Pain, V.M. Functional analysis of individual binding activities of the scaffold protein eIF4G. J. Biol. Chem. 2007, 282, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.-S.; Kim, J.-R.; Walker, S.E.; Dong, J.; Lorsch, J.R.; Dever, T.E. Initiation factor eIF2γ promotes eIF2–GTP–Met-tRNAMet ternary complex binding to the 40S ribosome. Nat. Struct. Mol. Biol. 2011, 18, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Methot, N.; Song, M.S.; Sonenberg, N. A region rich in aspartic acid, arginine, tyrosine, and glycine (DRYG) mediates eukaryotic initiation factor 4B (eIF4B) self-association and interaction with eIF3. Mol. Cell Biol. 1996, 16, 5328–5334. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Lorsch, J.R. The mechanism of eukaryotic translation initiation: New insights and challenges. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Clayton, J.; Shalev, A.; Hinnebusch, A.G. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000, 14, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Valásek, L.; Nielsen, K.H.; Zhang, F.; Fekete, C.A.; Hinnebusch, A.G. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 2004, 24, 9437–9455. [Google Scholar] [CrossRef] [PubMed]
- Rozovsky, N.; Butterworth, A.C.; Moore, M.J. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA 2008, 14, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- LeFebvre, A.K.; Korneeva, N.L.; Trutschl, M.; Cvek, U.; Duzan, R.D.; Bradley, C.A.; Hershey, J.W.B.; Rhoads, R.E. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3E subunit. J. Biol. Chem. 2006, 281, 22917–22932. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Influence of mRNA secondary structure on binding and migration of 40S ribosomal subunits. Cell 1980, 19, 79–90. [Google Scholar] [CrossRef]
- Parsyan, A.; Svitkin, Y.; Shahbazian, D.; Gkogkas, C.; Lasko, P.; Merrick, W.C.; Sonenberg, N. MRNA helicases: The tacticians of translational control. Nat. Rev. Mol. Cell Biol. 2011, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, K.; Muldoon, M.; Rajkowitsch, L.; Hughes, J.; McCarthy, J.E.G. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 2004, 51, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Parsyan, A.; Shahbazian, D.; Martineau, Y.; Petroulakis, E.; Alain, T.; Larsson, O.; Mathonnet, G.; Tettweiler, G.; Hellen, C.U.; Pestova, T.V.; et al. The helicase protein DHX29 promotes translation initiation, cell proliferation, and tumorigenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 22217–22222. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Dias, A.P.; Jedrychowski, M.; Patel, A.H.; Hsu, J.L.; Reed, R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 2008, 36, 4708–4718. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Qian, S.; Bolinger, C.; Fernandez, S.; Schoenberg, D.R.; Boris-Lawrie, K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 2006, 13, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Pisareva, V.P.; Pisarev, A.V.; Komar, A.A.; Hellen, C.U.T.; Pestova, T.V. Translation initiation on mammalian mRNAs with structured 5′UTRs requires DExH-box protein DHX29. Cell 2008, 135, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Lomakin, I.B.; Lee, J.H.; Choi, S.K.; Dever, T.E.; Hellen, C.U. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 2000, 403, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Kräusslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [PubMed]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J. Alternative mechanisms of initiating translation of mammalian mRNAs. Biochem. Soc. Trans. 2005, 33, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.R.; Abaeva, I.S.; Pestova, T.V.; Hellen, C.U.T. The mechanism of translation initiation on type 1 picornavirus IRESs. EMBO J. 2014, 33, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Schüler, M.; Connell, S.R.; Lescoute, A.; Giesebrecht, J.; Dabrowski, M.; Schroeer, B.; Mielke, T.; Penczek, P.A.; Westhof, E.; Spahn, C.M.T. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 2006, 13, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Spahn, C.M.T.; Jan, E.; Mulder, A.; Grassucci, R.A.; Sarnow, P.; Frank, J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: The IRES functions as an RNA-based translation factor. Cell 2004, 118, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Kieft, J.S. Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci. 2008, 33, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Sharp, P.A.; Jamison, S.F.; Garcia-Blanco, M.A. Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev. 1991, 5, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.A.; Spriggs, K.A.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. The APAF-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and UNR. Mol. Cell 2003, 11, 757–771. [Google Scholar] [CrossRef]
- Izumi, R.E.; Das, S.; Barat, B.; Raychaudhuri, S.; Dasgupta, A. A peptide from autoantigen La blocks poliovirus and hepatitis C virus cap-independent translation and reveals a single tyrosine critical for La RNA binding and translation stimulation. J. Virol. 2004, 78, 3763–3776. [Google Scholar] [CrossRef] [PubMed]
- McBratney, S.; Sarnow, P. Evidence for involvement of trans-acting factors in selection of the AUG start codon during eukaryotic translational initiation. Mol. Cell. Biol. 1996, 16, 3523–3534. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, A.; Hunt, S.L.; Patton, J.G.; Jackson, R.J. Direct evidence that polypyrimidine tract binding protein (PTB) is essential for internal initiation of translation of encephalomyocarditis virus RNA. RNA 1995, 1, 924–938. [Google Scholar] [PubMed]
- Costa-Mattioli, M.; Svitkin, Y.; Sonenberg, N. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 6861–6870. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.D.; Semler, B.L. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 2009, 1789, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Meerovitch, K.; Lee, H.S.; Dholakia, J.N.; Kenan, D.J.; Agol, V.I.; Sonenberg, N. Internal translation initiation on poliovirus RNA: Further characterization of La function in poliovirus translation in vitro. J. Virol. 1994, 68, 1544–1550. [Google Scholar] [PubMed]
- Hunt, S.L.; Jackson, R.J. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA 1999, 5, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.A.; Kong, X.; Haystead, T.A.; Pause, A.; Belsham, G.; Sonenberg, N.; Lawrence, J.C. Phas-I as a link between mitogen-activated protein kinase and translation initiation. Science 1994, 266, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, H.B.; Fumagalli, S.; Dennis, P.B.; Reinhard, C.; Pearson, R.B.; Thomas, G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997, 16, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two tor complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Helliwell, S.B.; Wagner, P.; Kunz, J.; Deuter-Reinhard, M.; Henriquez, R.; Hall, M.N. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell 1994, 5, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and RAPTOR-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C. Many roads from mTOR to eIF4F. Biochem. Soc. Trans. 2013, 41, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donzé, O.; Lin, T.A.; Lawrence, J.C.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-CAP function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Fadden, P.; Haystead, T.A.; Lawrence, J.C. Identification of phosphorylation sites in the translational regulator, phas-I that are controlled by insulin and rapamycin in rat adipocytes. J. Biol. Chem. 1997, 272, 10240–10247. [Google Scholar] [PubMed]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.B.; Blenis, J.; Pende, M.; Sonenberg, N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.P.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. MTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Ozes, A.R.; Feoktistova, K.; Avanzino, B.C.; Fraser, C.S. Duplex unwinding and atpase activities of the dead-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 2011, 412, 674–687. [Google Scholar] [PubMed]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Proud, C.G. Activation of mRNA translation in rat cardiac myocytes by insulin involves multiple rapamycin-sensitive steps. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1056–H1068. [Google Scholar] [PubMed]
- Redpath, N.T.; Foulstone, E.J.; Proud, C.G. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J. 1996, 15, 2291–2297. [Google Scholar] [PubMed]
- Gao, X.; Pan, D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001, 15, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/tor signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/AKT pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Algire, M.A.; Maag, D.; Lorsch, J.R. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 2005, 20, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Flynn, A.; Oldfield, S.; Proud, C.G. The role of the beta-subunit of initiation factor eIF-2 in initiation complex formation. Biochim. Biophys. Acta 1993, 1174, 117–121. [Google Scholar] [CrossRef]
- Pavitt, G.D.; Ramaiah, K.V.; Kimball, S.R.; Hinnebusch, A.G. EIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev. 1998, 12, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, T.; Pavitt, G.D.; Zhang, F.; Dever, T.E.; Hinnebusch, A.G. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 2001, 21, 5018–5030. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.R.; Udagawa, T.; Lee, B.; Wassink, S.; He, H.; Yamamoto, Y.; Anderson, J.T.; Pavitt, G.D.; Asano, K. Change in nutritional status modulates the abundance of critical pre-initiation intermediate complexes during translation initiation in vivo. J. Mol. Biol. 2007, 370, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.M.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef]
- Dever, T.E.; Hinnebusch, A.G. GCN2 whets the appetite for amino acids. Mol. Cell 2005, 18, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.R.; Zhang, F.; Tan, S.L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 n-terminal domain. Mol. Cell Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Perez, C.; Notary, J.; Mohr, I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 2005, 79, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 7029–7036. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.; Perez, C.; Mohr, I. Poly(A) binding protein abundance regulates eukaryotic translation initiation factor 4F assembly in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2012, 109, 5627–5632. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 2004, 78, 11030–11039. [Google Scholar] [CrossRef] [PubMed]
- Lenarcic, E.M.; Ziehr, B.; De Leon, G.; Mitchell, D.; Moorman, N.J. Differential role for host translation factors in host and viral protein synthesis during human cytomegalovirus infection. J. Virol. 2014, 88, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Shenk, T. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 2010, 84, 5260–5269. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.; Zavadil, J.; Bianco, C.; Shiflett, L.; Brown, S.; Mohr, I. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep. 2014, 6, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of RAPTOR- and RICTOR-containing complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 14182–14187. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Del Prete, G.Q.; Maguire, T.G.; Alwine, J.C. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J. Virol. 2007, 81, 3649–3651. [Google Scholar] [CrossRef] [PubMed]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J. Virol. 2011, 85, 12585–12593. [Google Scholar] [CrossRef] [PubMed]
- McArdle, J.; Moorman, N.J.; Munger, J. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 2012, 8, e1002502. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Xuan, B.; Liu, H.; Zhong, J.; Yu, D.; Qian, Z. Tuberous sclerosis complex protein 2-independent activation of mTORC1 by human cytomegalovirus pUL38. J. Virol. 2015, 89, 7625–7635. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.A.; Wang, X.; Ma, X.-L.; Huong, S.-M.; Huang, E.-S. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: Inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 2001, 75, 6022–6032. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The torrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-AKT-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J. Virol. 2011, 85, 3930–3939. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Schafer, X.L.; Moorman, N.J.; Munger, J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011, 85, 5814–5824. [Google Scholar] [CrossRef] [PubMed]
- Grainger, L.; Cicchini, L.; Rak, M.; Petrucelli, A.; Fitzgerald, K.D.; Semler, B.L.; Goodrum, F. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 2010, 84, 9472–9486. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, A.; Rak, M.; Grainger, L.; Goodrum, F. Characterization of a novel golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 2009, 83, 5615–5629. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pierciey, F.J.; Maguire, T.G.; Alwine, J.C. PKR-like endoplasmic reticulum kinase is necessary for lipogenic activation during HCMV infection. PLoS Pathog. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Maguire, T.G.; Yu, Y.; Paton, A.W.; Paton, J.C.; Alwine, J.C. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 2008, 82, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Pierciey, F.J.; Alwine, J.C. Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site. J. Virol. 2010, 84, 11479–11486. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Back, S.H.; Rho, J.; Lee, S.H.; Jang, S.K. La autoantigen enhances translation of BiP mRNA. Nucleic Acids Res. 2001, 29, 5009–5016. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TSR1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Hakki, M.; De Niro, K.L.; Geballe, A.P. Evasion of cellular antiviral responses by human cytomegalovirus TSR1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Romanowski, M.J.; Shenk, T. Characterization of the human cytomegalovirus IRS1 and TRS1 genes: A second immediate-early transcription unit within IRS1 whose product antagonizes transcriptional activation. J. Virol. 1997, 71, 1485–1496. [Google Scholar] [PubMed]
- Hakki, M.; Geballe, A.P. Double-stranded RNA binding by human cytomegalovirus PTRS1. J. Virol. 2005, 79, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Bierle, C.J.; Semmens, K.M.; Geballe, A.P. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 2013, 442, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed]
- Braggin, J.E.; Child, S.J.; Geballe, A.P. Essential role of protein kinase R antagonism by TRS1 in human cytomegalovirus replication. Virology 2016, 489, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human cytomegalovirus PTRS1 and PIRS1 antagonize PKR to facilitate virus replication. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Geballe, A.P. Cellular serine/threonine phosphatase activity during human cytomegalovirus infection. Virology 2008, 380, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, O.; Cohen, Y.; Shitrit, A.; Shani, O.; Le-Trilling, V.T.; Trilling, M.; Friedlander, G.; Tanenbaum, M.; Stern-Ginossar, N. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog. 2015, 11, e1005288. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.K.; Hein, M.Y.; Huang, S.-X.; Ma, M.; Shen, B.; Qian, S.-B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd; Kouzarides, T.; Martignetti, J.A.; et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar] [PubMed]
- Murphy, E.; Shenk, T. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 2008, 325, 1–19. [Google Scholar] [PubMed]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Yachie, N.; Okada, Y.; Saito, R.; Tomita, M. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett. 2007, 581, 4184–4188. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Watatani, Y.; Ichikawa, K.; Nakanishi, N.; Fujimoto, M.; Takeda, H.; Kimura, N.; Hirose, H.; Takahashi, S.; Takahashi, Y. Stress-induced translation of ATF5 mRNA is regulated by the 5′-untranslated region. J. Biol. Chem. 2008, 283, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Raghavan, B.; Kotur, M.; Cheatham, J.; Sedmak, D.; Cook, C.; Waldman, J.; Trgovcich, J. Antisense transcription in the human cytomegalovirus transcriptome. J. Virol. 2007, 81, 11267–11281. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, N.; Li, M.; Gao, S.; Wang, L.; Zheng, B.; Qi, Y.; Ruan, Q. Human CMV transcripts: An overview. Future Microbiol. 2012, 7, 577–593. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, H.A.; Ziehr, B.; Moorman, N.J. Human Cytomegalovirus Strategies to Maintain and Promote mRNA Translation. Viruses 2016, 8, 97. https://doi.org/10.3390/v8040097
Vincent HA, Ziehr B, Moorman NJ. Human Cytomegalovirus Strategies to Maintain and Promote mRNA Translation. Viruses. 2016; 8(4):97. https://doi.org/10.3390/v8040097
Chicago/Turabian StyleVincent, Heather A., Benjamin Ziehr, and Nathaniel J. Moorman. 2016. "Human Cytomegalovirus Strategies to Maintain and Promote mRNA Translation" Viruses 8, no. 4: 97. https://doi.org/10.3390/v8040097
APA StyleVincent, H. A., Ziehr, B., & Moorman, N. J. (2016). Human Cytomegalovirus Strategies to Maintain and Promote mRNA Translation. Viruses, 8(4), 97. https://doi.org/10.3390/v8040097