
Conserved Endonuclease Function of Hantavirus L Polymerase

Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling
2.2. Construction of Plasmids
2.3. Cells
2.4. Reporter Assays
2.5. Western Blotting
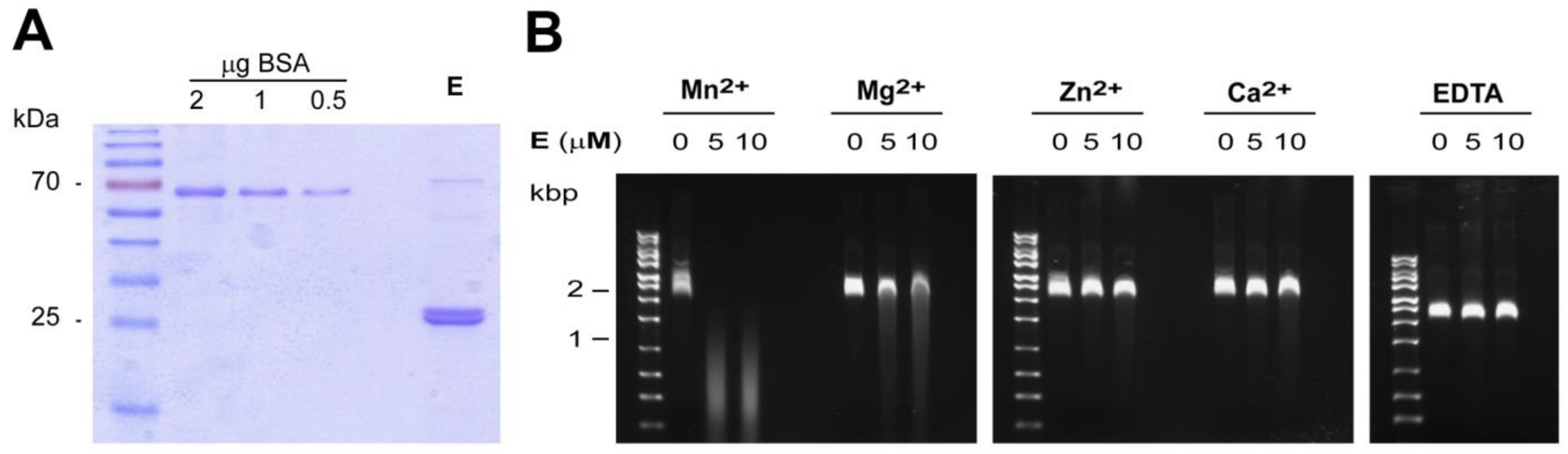
2.6. Protein Expression and Purification
2.7. In Vitro Endonuclease Assay
2.8. Cell Viability
2.9. Statistical Analysis
3. Results
3.1. Conservation of the N-Terminal Endonuclease of Hantavirus L Polymerase
3.2. In Vitro Activity and Cation-Dependence of HTNV L Endonuclease
3.3. Development of a Cell-Based Assay for Hantavirus Endonuclease
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walter, C.T.; Barr, J.N. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 2011, 92, 2467–2484. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Brennan, B. Emerging phleboviruses. Curr. Opin. Virol. 2014, 5, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M. Orthobunyaviruses: Recent genetic and structural insights. Nat. Rev. Microbiol. 2014, 12, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Manigold, T.; Vial, P. Human hantavirus infections: Epidemiology, clinical features, pathogenesis and immunology. Schweiz. Med. Wochenschr. 2014, 144, w13937. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.C.; Sargianou, M.; Papa, A.; Chra, P.; Starakis, I.; Panos, G. Epidemiology of Hantavirus infections in humans: A comprehensive, global overview. Crit. Rev. Microbiol. 2014, 40, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Heyman, P.; Ceianu, C.S.; Christova, I.; Tordo, N.; Beersma, M.; Joao Alves, M.; Lundkvist, A.; Hukic, M.; Papa, A.; Tenorio, A.; et al. A five-year perspective on the situation of haemorrhagic fever with renal syndrome and status of the hantavirus reservoirs in Europe, 2005–2010. Euro Surveill. 2011, 16, 15–22. [Google Scholar]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.H.; Schonrich, G.; Klempa, B. Human pathogenic hantaviruses and prevention of infection. Hum. Vaccines 2011, 7, 685–693. [Google Scholar] [CrossRef]
- Macneil, A.; Nichol, S.T.; Spiropoulou, C.F. Hantavirus pulmonary syndrome. Virus Res. 2011, 162, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Bahr, U.; Zeier, M.; van der Woude, F.J. Hantavirus infection. J. Am. Soc. Nephrol. 2005, 16, 3669–3679. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.S.; Nichol, S.T. Bunyaviridae. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1741–1790. [Google Scholar]
- Papa, A. Dobrava-Belgrade virus: Phylogeny, epidemiology, disease. Antivir. Res. 2012, 95, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Makela, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, S.K.; Vaheri, A.; Plyusnin, A. L protein, the RNA-dependent RNA polymerase of hantaviruses. Arch. Virol. 2005, 150, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Plotch, S.J.; Bouloy, M.; Krug, R.M. Transfer of 5′-terminal cap of globin mRNA to influenza viral complementary RNA during transcription in vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Steczkiewicz, K.; Muszewska, A.; Knizewski, L.; Rychlewski, L.; Ginalski, K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012, 40, 7016–7045. [Google Scholar] [CrossRef] [PubMed]
- Crepin, T.; Dias, A.; Palencia, A.; Swale, C.; Cusack, S.; Ruigrok, R.W. Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J. Virol. 2010, 84, 9096–9104. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.H.; Gay, M.E.; Matsuoko, Y. Nonviral heterogeneous sequences are present at the 5′ ends of one species of snowshoe hare bunyavirus S complementary RNA. Nucleic Acids Res. 1983, 11, 6409–6418. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Weber, F.; Cusack, S. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010, 6, e1001101. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, P.; Schmidt-Chanasit, J.; Gunther, S. The N terminus of Andes virus L protein suppresses mRNA and protein expression in mammalian cells. J. Virol. 2013, 87, 6975–6985. [Google Scholar] [CrossRef] [PubMed]
- DuBois, R.M.; Slavish, P.J.; Baughman, B.M.; Yun, M.K.; Bao, J.; Webby, R.J.; Webb, T.R.; White, S.W. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog. 2012, 8, e1002830. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007. Chapter 2, Unit 2.9. [Google Scholar] [CrossRef]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Flick, K.; Hooper, J.W.; Schmaljohn, C.S.; Pettersson, R.F.; Feldmann, H.; Flick, R. Rescue of Hantaan virus minigenomes. Virology 2003, 306, 219–224. [Google Scholar] [CrossRef]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [PubMed]
- Klemm, C.; Reguera, J.; Cusack, S.; Zielecki, F.; Kochs, G.; Weber, F. Systems to establish bunyavirus genome replication in the absence of transcription. J. Virol. 2013, 87, 8205–8212. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.S. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bridgen, A.; Elliott, R.M. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc. Natl. Acad. Sci. USA 1996, 93, 15400–15404. [Google Scholar] [CrossRef] [PubMed]
- Rezelj, V.V.; Overby, A.K.; Elliott, R.M. Generation of mutant Uukuniemi viruses lacking the nonstructural protein NSs by reverse genetics indicates that NSs is a weak interferon antagonist. J. Virol. 2015, 89, 4849–4856. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.; Li, P.; Zhang, S.; Li, A.; Liang, M.; Li, D.; Elliott, R.M. Reverse genetics system for severe fever with thrombocytopenia syndrome virus. J. Virol. 2015, 89, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rescue of infectious rift valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol. 2006, 80, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, E.; Albarino, C.G.; Khristova, M.L.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J. Virol. 2010, 84, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, E.; Zivcec, M.; Chakrabarti, A.K.; Nichol, S.T.; Albarino, C.G.; Spiropoulou, C.F. Recovery of Recombinant Crimean Congo Hemorrhagic Fever Virus Reveals a Function for Non-structural Glycoproteins Cleavage by Furin. PLoS Pathog. 2015, 11, e1004879. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.S.; Ebihara, H.; Feldmann, H. Development of a minigenome system for Andes virus, a New World hantavirus. Arch. Virol. 2012, 157, 2227–2233. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, P.; Malet, H.; Cusack, S.; Reguera, J. Structural Insights into Bunyavirus Replication and Its Regulation by the vRNA Promoter. Cell 2015, 161, 1267–1279. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | PUUV | HTNV |
|---|---|---|
| Number 1 | 0.94 | 0.93 |
| Number 2 | 0.93 | 0.90 |
| Number 3 | 0.94 | 0.90 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothenberger, S.; Torriani, G.; Johansson, M.U.; Kunz, S.; Engler, O. Conserved Endonuclease Function of Hantavirus L Polymerase. Viruses 2016, 8, 108. https://doi.org/10.3390/v8050108
Rothenberger S, Torriani G, Johansson MU, Kunz S, Engler O. Conserved Endonuclease Function of Hantavirus L Polymerase. Viruses. 2016; 8(5):108. https://doi.org/10.3390/v8050108
Chicago/Turabian StyleRothenberger, Sylvia, Giulia Torriani, Maria U. Johansson, Stefan Kunz, and Olivier Engler. 2016. "Conserved Endonuclease Function of Hantavirus L Polymerase" Viruses 8, no. 5: 108. https://doi.org/10.3390/v8050108
APA StyleRothenberger, S., Torriani, G., Johansson, M. U., Kunz, S., & Engler, O. (2016). Conserved Endonuclease Function of Hantavirus L Polymerase. Viruses, 8(5), 108. https://doi.org/10.3390/v8050108