
Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies and Reagents
2.3. MTT Assay
2.4. CHIKV Infection in Macrophage
2.5. Plaque Assay
2.6. Flow Cytometry (FC)
2.7. Sandwich ELISA for Cytokine Analysis
2.8. Annexin V Staining
2.9. Western Blot Analysis
2.10. RT-PCR
2.11. Statistical Analysis
3. Results
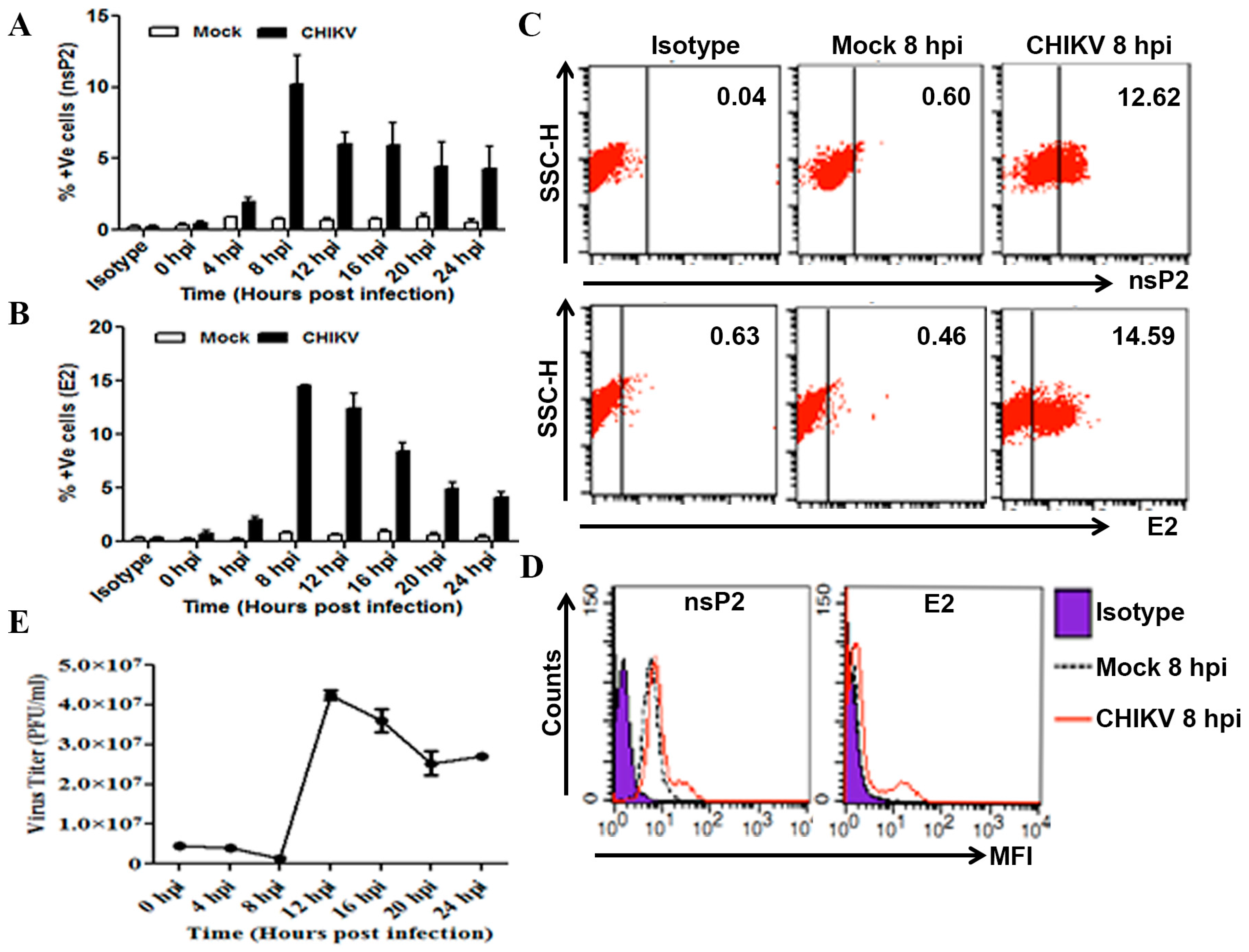
3.1. Determination of CHIKV Infection in Macrophages
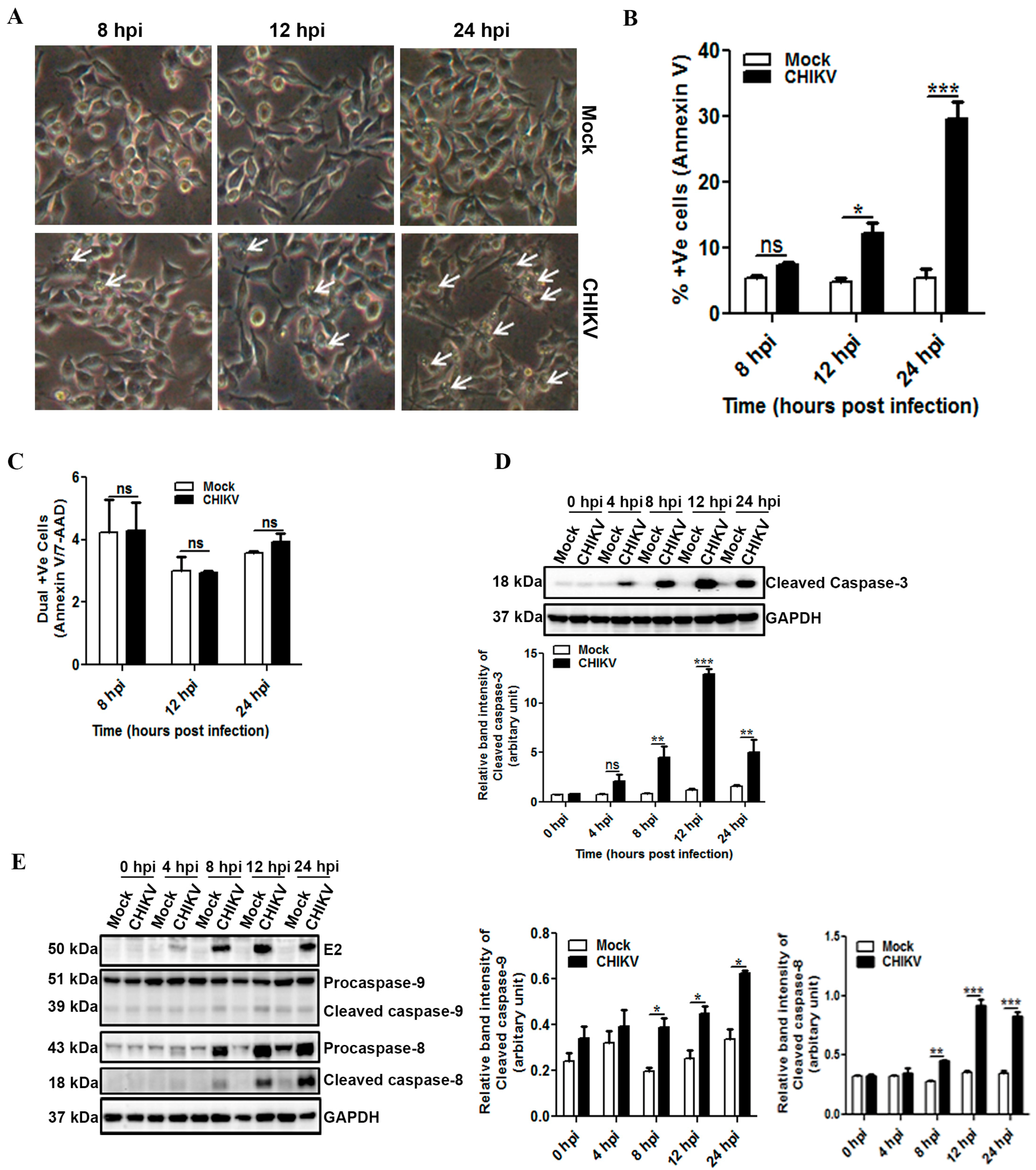
3.2. CHIKV Infection Induces Apoptosis in Macrophages
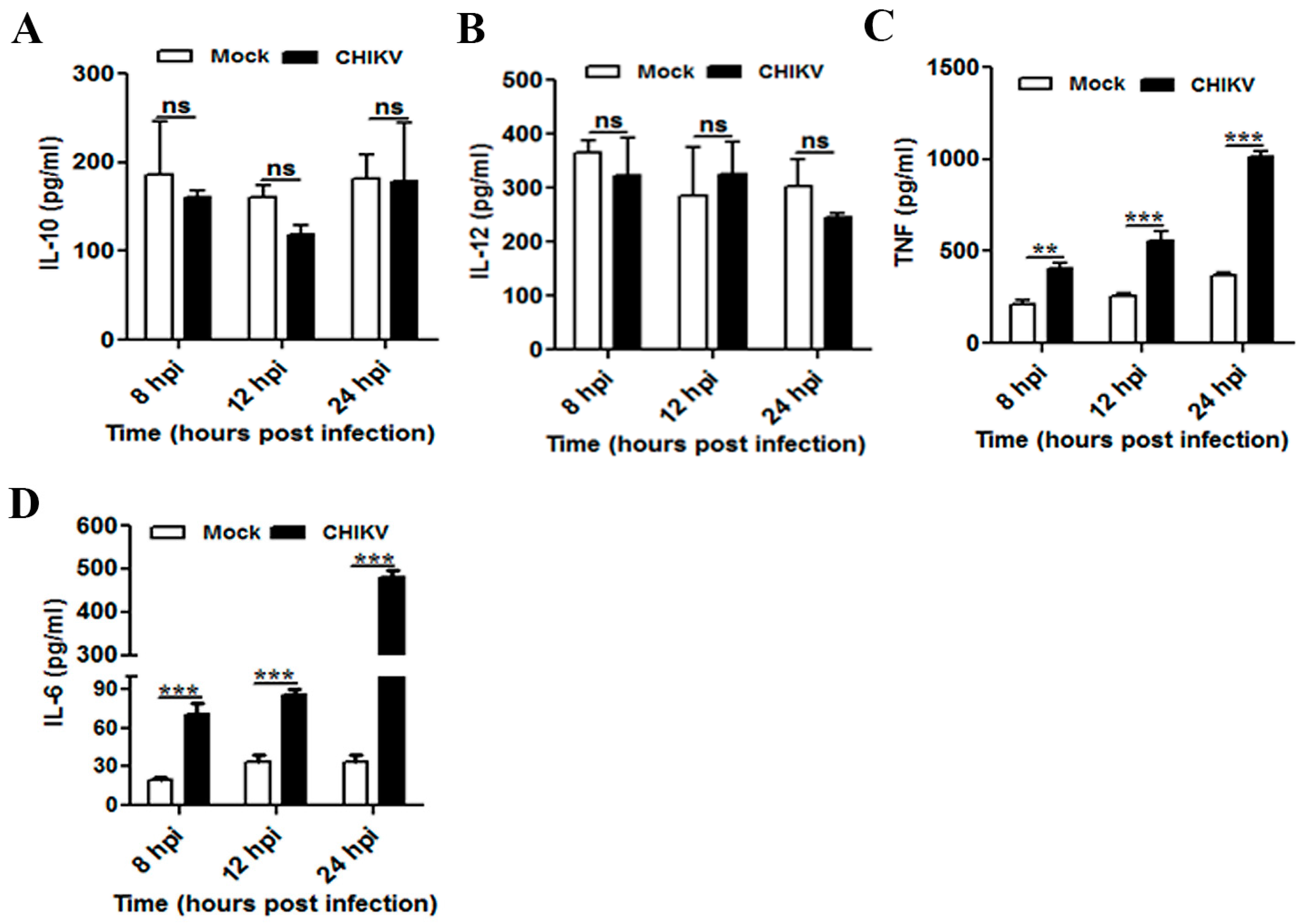
3.3. CHIKV Infection Upregulates Pro-Inflammatory Response in Mouse Macrophages
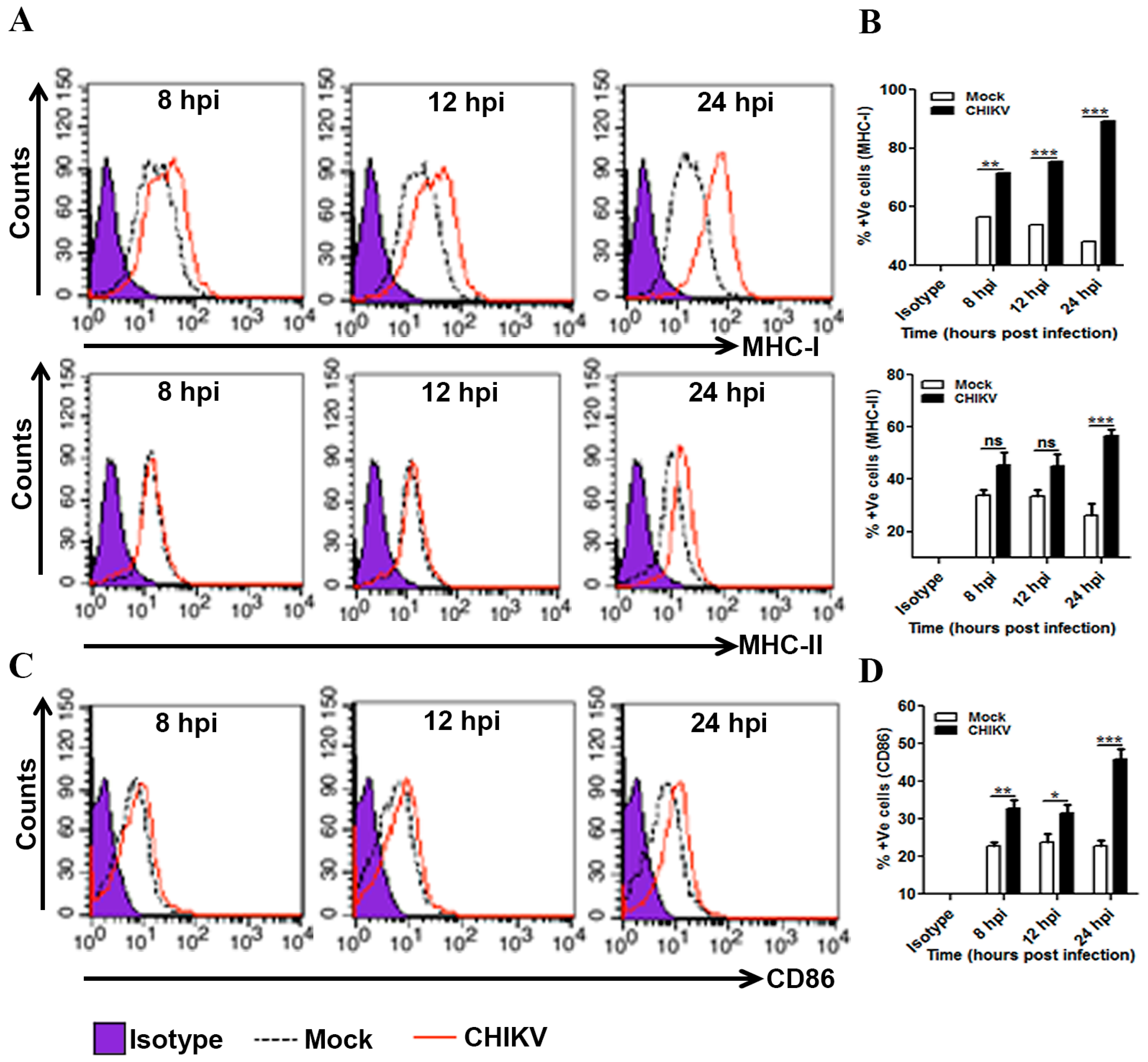
3.4. Induction of MHCs and Co-Stimulatory Molecules in CHIKV Infected Macrophages
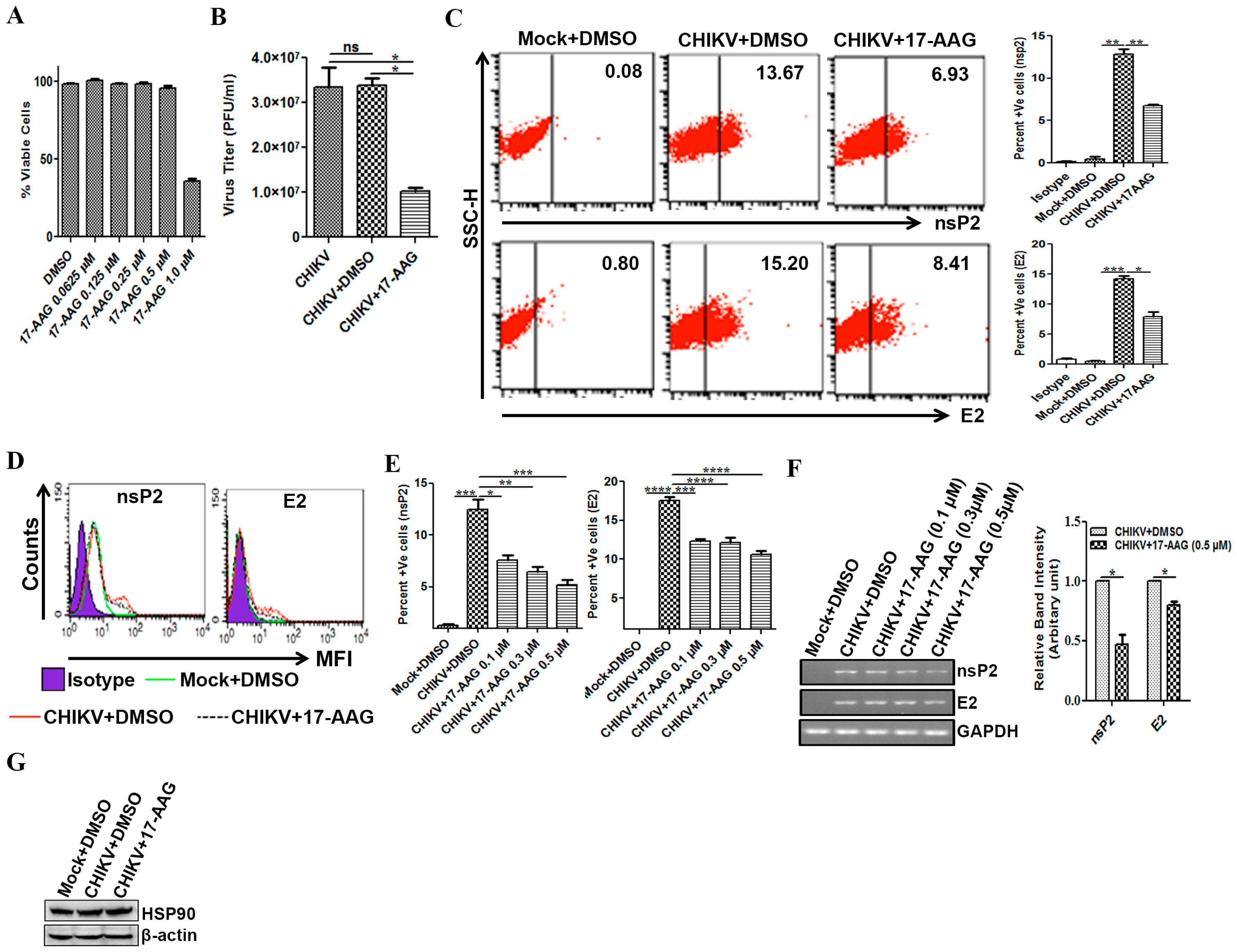
3.5. Regulation of CHIKV Infection by 17-AAG in Macrophages
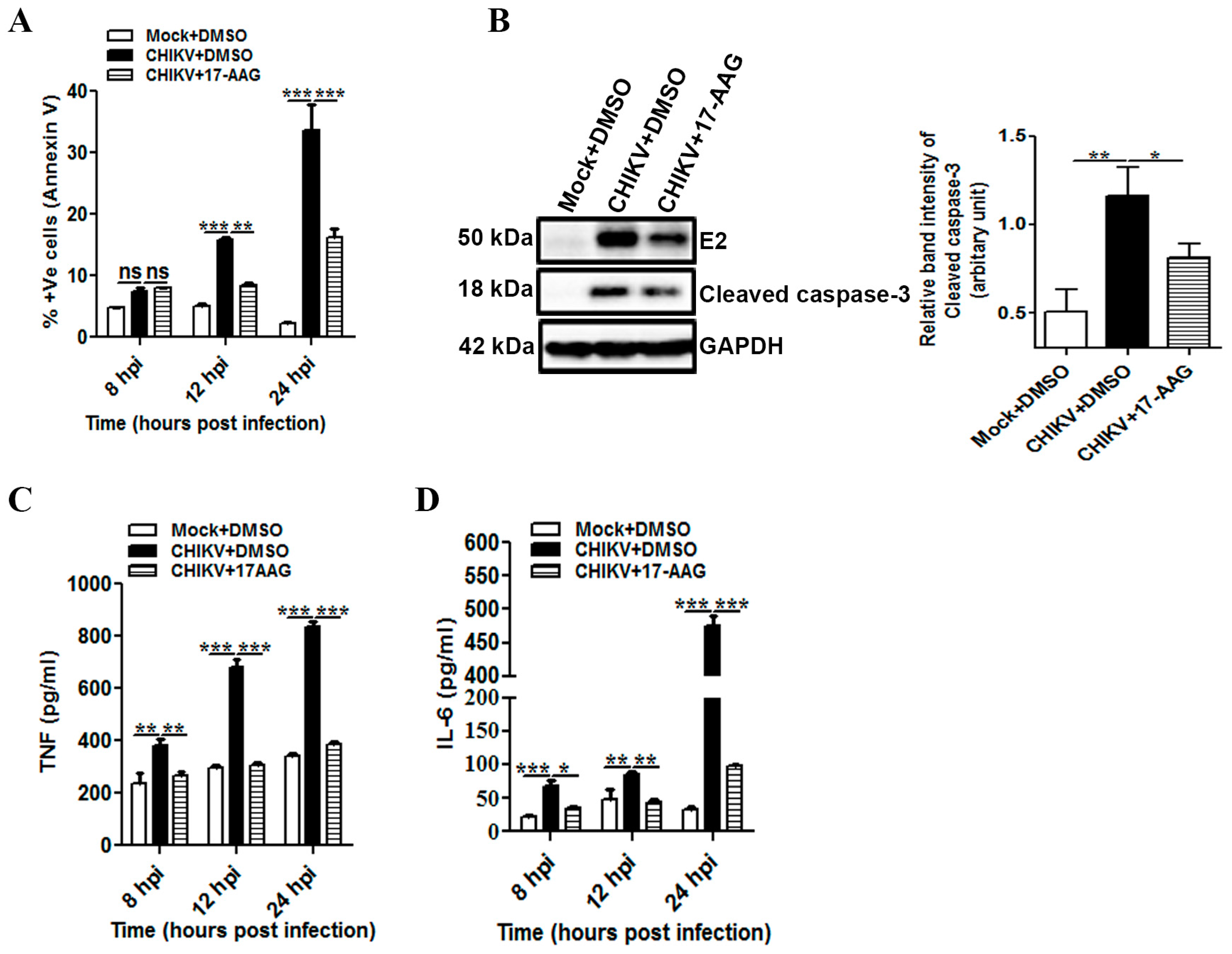
3.6. 17-AAG Regulates Apoptosis and Cellular Immune Responses during CHIKV Infection in Macrophages
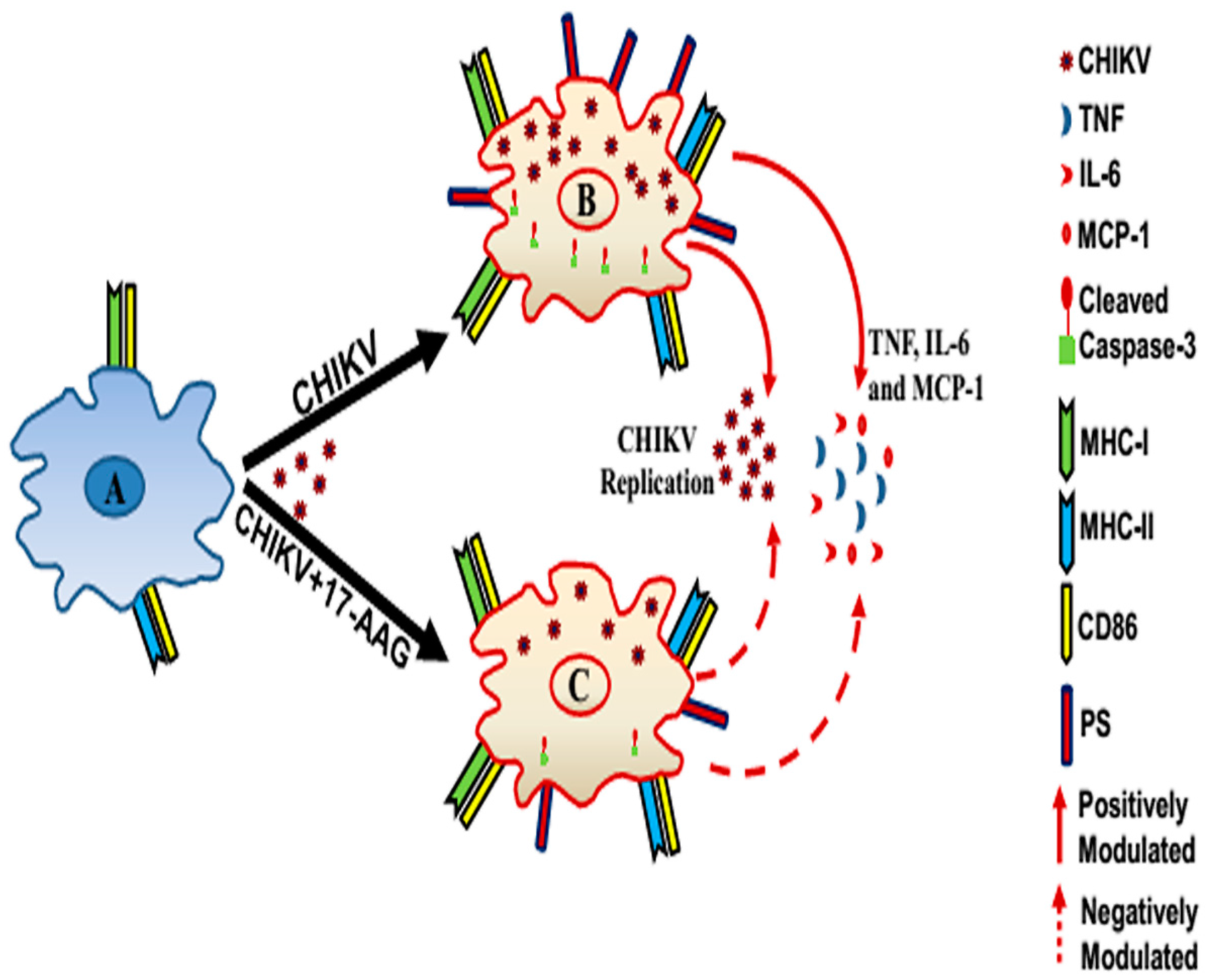
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khatun, S.; Chakraborty, A.; Rahman, M.; Nasreen Banu, N.; Rahman, M.M.; Hasan, S.M.; Luby, S.P.; Gurley, E.S. An outbreak of chikungunya in rural bangladesh, 2011. PLoS Negl. Trop. Dis. 2015, 9, e0003907. [Google Scholar] [CrossRef] [PubMed]
- Kosasih, H.; de Mast, Q.; Widjaja, S.; Sudjana, P.; Antonjaya, U.; Ma’roef, C.; Riswari, S.F.; Porter, K.R.; Burgess, T.H.; Alisjahbana, B.; et al. Evidence for endemic chikungunya virus infections in bandung, indonesia. PLoS Negl. Trop. Dis. 2013, 7, e2483. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Logue, C.H. Changing patterns of chikungunya virus: Re-emergence of a zoonotic arbovirus. J. Gen. Virol. 2007, 88, 2363–2377. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Barraquer, I.; Solomon, S.S.; Kuganantham, P.; Srikrishnan, A.K.; Vasudevan, C.K.; Iqbal, S.H.; Balakrishnan, P.; Solomon, S.; Mehta, S.H.; Cummings, D.A. The hidden burden of dengue and chikungunya in chennai, India. PLoS Negl. Trop. Dis. 2015, 9, e0003906. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.C. An epidemic of virus disease in southern province, tanganyika territory, in 1952–53. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. [Google Scholar] [CrossRef]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Thiberville, S.D.; Boisson, V.; Gaudart, J.; Simon, F.; Flahault, A.; de Lamballerie, X. Chikungunya fever: A clinical and virological investigation of outpatients on Reunion Island, South-West Indian Ocean. PLoS Negl. Trop. Dis. 2013, 7, e2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Zim, M.A.; Sam, I.C.; Omar, S.F.; Chan, Y.F.; AbuBakar, S.; Kamarulzaman, A. Chikungunya infection in Malaysia: Comparison with dengue infection in adults and predictors of persistent arthralgia. J. Clin. Virol. 2013, 56, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Borgherini, G.; Poubeau, P.; Staikowsky, F.; Lory, M.; le Moullec, N.; Becquart, J.P.; Wengling, C.; Michault, A.; Paganin, F. Outbreak of chikungunya on reunion island: Early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 2007, 44, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Maiti, C.R.; Mukherjee, A.K.; Bose, B.; Saha, G.L. Myopericarditis following Chikungunya virus infection. J. Indian Med. Assoc. 1978, 70, 256–258. [Google Scholar] [PubMed]
- Mittal, A.; Mittal, S.; Bharati, M.J.; Ramakrishnan, R.; Saravanan, S.; Sathe, P.S. Optic neuritis associated with Chikungunya virus infection in South India. Arch. Ophthalmol. 2007, 125, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, P.; Rathinam, S.; Banushree, K.; Maheshkumar, S.; Vijayakumar, R.; Sathe, P. Ocular involvement associated with an epidemic outbreak of chikungunya virus infection. Am. J. Ophthalmol. 2007, 144, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Solanki, B.S.; Arya, S.C.; Maheshwari, P. Chikungunya disease with nephritic presentation. Int. J. Clin. Pract. 2007, 61. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Paule, P.; Oliver, M. Chikungunya virus-induced myopericarditis: Toward an increase of dilated cardiomyopathy in countries with epidemics? Am. J. Trop. Med. Hyg. 2008, 78, 212–213. [Google Scholar] [PubMed]
- Chusri, S.; Siripaitoon, P.; Hirunpat, S.; Silpapojakul, K. Case reports of neuro-Chikungunya in Southern Thailand. Am. J. Trop. Med. Hyg. 2011, 85, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Renault, P.; Solet, J.L.; Sissoko, D.; Balleydier, E.; Larrieu, S.; Filleul, L.; Lassalle, C.; Thiria, J.; Rachou, E.; de Valk, H.; et al. A major epidemic of Chikungunya virus infection on reunion island, France, 2005–2006. Am. J. Trop. Med. Hyg. 2007, 77, 727–731. [Google Scholar] [PubMed]
- Thiboutot, M.M.; Kannan, S.; Kawalekar, O.U.; Shedlock, D.J.; Khan, A.S.; Sarangan, G.; Srikanth, P.; Weiner, D.B.; Muthumani, K. Chikungunya: A potentially emerging epidemic? PLoS Negl. Trop. Dis. 2010, 4, e623. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Liu, W.J.; Prow, N.A.; Geertsema, C.; Ligtenberg, M.; Vanlandingham, D.L.; Schnettler, E.; Vlak, J.M.; Suhrbier, A.; Khromykh, A.A.; et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 2010, 84, 10877–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelvin, A.A.; Banner, D.; Silvi, G.; Moro, M.L.; Spataro, N.; Gaibani, P.; Cavrini, F.; Pierro, A.; Rossini, G.; Cameron, M.J.; et al. Inflammatory cytokine expression is associated with Chikungunya virus resolution and symptom severity. PLoS Negl. Trop. Dis. 2011, 5, e1279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jaffar-Bandjee, M.C.; Giry, C.; Connen de Kerillis, L.; Merits, A.; Gasque, P.; Hoarau, J.J. Mouse macrophage innate immune response to Chikungunya virus infection. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Dhanwani, R.; Patro, I.K.; Rao, P.V.; Parida, M.M. Differential regulation of TLR mediated innate immune response of mouse neuronal cells following infection with novel ECSA genotype of Chikungunya virus with and without E1:A226V mutation. Infect. Genet. Evol. 2013, 20, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Patro, I.K.; Parida, M.M. TLR3 mediated innate immune response in mice brain following infection with Chikungunya virus. Virus Res. 2014, 189, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Chirathaworn, C.; Poovorawan, Y.; Lertmaharit, S.; Wuttirattanakowit, N. Cytokine levels in patients with Chikungunya virus infection. Asian Pac. J. Trop. Med. 2013, 6, 631–634. [Google Scholar] [CrossRef]
- Chow, A.; Her, Z.; Ong, E.K.; Chen, J.M.; Dimatatac, F.; Kwek, D.J.; Barkham, T.; Yang, H.; Renia, L.; Leo, Y.S.; et al. Persistent arthralgia induced by Chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J. Infect. Dis. 2011, 203, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lohachanakul, J.; Phuklia, W.; Thannagith, M.; Thongsakulprasert, T.; Smith, D.R.; Ubol, S. Differences in response of primary human myoblasts to infection with recent epidemic strains of Chikungunya virus isolated from patients with and without myalgia. J. Med. Virol. 2015, 87, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Phuklia, W.; Kasisith, J.; Modhiran, N.; Rodpai, E.; Thannagith, M.; Thongsakulprasert, T.; Smith, D.R.; Ubol, S. Osteoclastogenesis induced by CHIKV-infected fibroblast-like synoviocytes: A possible interplay between synoviocytes and monocytes/macrophages in CHIKV-induced arthralgia/arthritis. Virus Res. 2014, 177, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Mani, R.S.; Desai, A.; Ravi, V. Correlation of plasma viral loads and presence of Chikungunya igm antibodies with cytokine/chemokine levels during acute Chikungunya virus infection. J. Med. Virol. 2014, 86, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Chow, A.; Sun, Y.J.; Kwek, D.J.; Lim, P.L.; Dimatatac, F.; Ng, L.C.; Ooi, E.E.; Choo, K.H.; Her, Z.; et al. Il-1beta, IL-6, and rantes as biomarkers of Chikungunya severity. PLoS ONE 2009, 4, e4261. [Google Scholar] [CrossRef] [PubMed]
- Daley-Bauer, L.P.; Wynn, G.M.; Mocarski, E.S. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity 2012, 37, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Pratheek, B.M.; Saha, S.; Maiti, P.K.; Chattopadhyay, S.; Chattopadhyay, S. Immune regulation and evasion of mammalian host cell immunity during viral infection. Indian J. Virol. 2013, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Yao, J.; Yang, F.; Nie, J.; Wang, Y.; Liu, P.C. Human immunodeficiency virus type 1 Nef in human monocyte-like cell line THP-1 expands treg cells via toll-like receptor 2. J. Cell. Biochem. 2011, 112, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Parga, T.; Sehrawat, S.; Rouse, B.T. Role of regulatory T cells during virus infection. Immunol. Rev. 2013, 255, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.; Wong, S.C.; Kwek, D.J.; Tolou, H.; Lin, R.T.; Tambyah, P.A.; Renia, L.; et al. Active infection of human blood monocytes by chikungunya virus triggers an innate immune response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef] [PubMed]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Invest. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, J.J.; Jaffar Bandjee, M.C.; Krejbich Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Rogon, C.; Hohfeld, J. To be, or not to be—Molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007, 64, 2525–2541. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Tam, S.; Kaganovich, D.; Frydman, J. Protein quality control: Chaperones culling corrupt conformations. Nat. Cell Biol. 2005, 7, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J. How cells respond to stress. Sci. Am. 1993, 268, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.; Ruby, J. Heat shock response to vaccinia virus infection. J. Virol. 1994, 68, 4685–4689. [Google Scholar] [PubMed]
- Sullivan, C.S.; Pipas, J.M. The virus-chaperone connection. Virology 2001, 287, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Basha, W.; Kitagawa, R.; Uhara, M.; Imazu, H.; Uechi, K.; Tanaka, J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir. Chem. Chemother. 2005, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Bagchi, P.; Chatterjee, A.; Nayak, M.K.; Mukherjee, A.; Chattopadhyay, S.; Nagashima, S.; Kobayashi, N.; Komoto, S.; Taniguchi, K.; et al. The molecular chaperone heat shock protein-90 positively regulates rotavirus infectionx. Virology 2009, 391, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Vignuzzi, M.; Andino, R.; Frydman, J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007, 21, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1996, 93, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Chung, C.S.; Chang, W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002, 76, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Barlow, E.A.; Ma, S.; Hagemeier, S.R.; Duellman, S.J.; Burgess, R.R.; Tellam, J.; Khanna, R.; Kenney, S.C. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.Z.; Miao, J.; Zhao, M.; Tang, M.; Yeo, A.E.; Yu, H.; Zhang, J.; Xia, N.S. Role of heat-shock protein 90 in hepatitis E virus capsid trafficking. J. Gen. Virol. 2010, 91, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Guzik, K.; Slezak, K.; Dziedzic, J.; Rokita, H. Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. J. Inflamm. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Basantray, I.; Mamidi, P.; Nayak, T.K.; Pratheek, B.M.; Chattopadhyay, S.; Chattopadhyay, S. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS ONE 2014, 9, e100531. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.P.; Haystead, T.; Das, P.K.; Merits, A.; Ng, M.L.; Vasudevan, S.G. Chikungunya virus nsP3 and nsP4 interacts with Hsp-90 to promote virus replication: Hsp-90 inhibitors reduce CHIKV infection and inflammation in vivo. Antivir. Res. 2014, 103, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Agnew, E.B.; Wilson, R.H.; Grem, J.L.; Neckers, L.; Bi, D.; Takimoto, C.H. Measurement of the novel antitumor agent 17-(allylamino)-17-demethoxygeldanamycin in human plasma by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2001, 755, 237–243. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Kumar, A.; Mamidi, P.; Nayak, T.K.; Das, I.; Chhatai, J.; Basantray, I.; Bramha, U.; Maiti, P.K.; Singh, S.; et al. Development and characterization of monoclonal antibody against non-structural protein-2 of Chikungunya virus and its application. J. Virol. Methods 2014, 199, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mamidi, P.; Das, I.; Nayak, T.K.; Kumar, S.; Chhatai, J.; Chattopadhyay, S.; Suryawanshi, A.R. A novel 2006 Indian outbreak strain of Chikungunya virus exhibits different pattern of infection as compared to prototype strain. PLoS ONE 2014, 9, e85714. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Mamidi, P.; Kumar, S.; Basantray, I.; Saswat, T.; Das, I.; Nayak, T.K.; Chattopadhyay, S.; Subudhi, B.B.; et al. Inhibition of chikungunya virus replication by 1-[(2-methylbenzimidazol-1-yl) methyl]-2-oxo-indolin-3-ylidene] amino] thiourea(mbzm-n-ibt). Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; le Roux, K.; Prevost, M.C.; Fsihi, H.; et al. Characterization of reemerging Chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, F.; Jaworska, J.; Verway, M.; Tzelepis, F.; Massoud, A.; Gillard, J.; Wong, G.; Kobinger, G.; Xing, Z.; Couture, C.; et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 2014, 40, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Krejbich-Trotot, P.; Denizot, M.; Hoarau, J.J.; Jaffar-Bandjee, M.C.; Das, T.; Gasque, P. Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. FASEB J. 2011, 25, 314–325. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhou, H.R.; Pestka, J.J. Mechanisms for ribotoxin-induced ribosomal RNA cleavage. Toxicol. Appl. Pharmacol. 2012, 265, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Foo, S.S.; Taylor, A.; Lulla, A.; Merits, A.; Hueston, L.; Forwood, M.R.; Walsh, N.C.; Sims, N.A.; Herrero, L.J.; et al. Bindarit, an inhibitor of monocyte chemotactic protein synthesis, protects against bone loss induced by Chikungunya virus infection. J. Virol. 2015, 89, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.R.; Chevalier, M.S.; Johnson, D.C. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 2003, 24, 278–285. [Google Scholar] [CrossRef]
- Khan, N.; Gowthaman, U.; Pahari, S.; Agrewala, J.N. Manipulation of costimulatory molecules by intracellular pathogens: Veni, vidi, vici!! PLoS Pathog. 2012, 8, e1002676. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to Hsp90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998, 42, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Sakoonwatanyoo, P.; Ubol, S.; Yoksan, S.; Smith, D.R. Chikungunya virus infection of cell lines: Analysis of the east, central and south african lineage. PLoS ONE 2012, 7, e31102. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Mudaliar, P.; Padmanabhan, A.; Sreekumar, E. Induction of cytopathogenicity in human glioblastoma cells by Chikungunya virus. PLoS ONE 2013, 8, e75854. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Lundberg, L.; Swales, D.; Waybright, N.; Pinkham, C.; Dinman, J.D.; Jacobs, J.L.; Kehn-Hall, K. Venezuelan equine encephalitis virus induces apoptosis through the unfolded protein response activation of EGR1. J. Virol. 2016, 90, 3558–3572. [Google Scholar] [CrossRef] [PubMed]
- Dhanwani, R.; Khan, M.; Bhaskar, A.S.; Singh, R.; Patro, I.K.; Rao, P.V.; Parida, M.M. Characterization of chikungunya virus infection in human neuroblastoma SH-SY5Y cells: Role of apoptosis in neuronal cell death. Virus Res. 2012, 163, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Way, S.J.; Lidbury, B.A.; Banyer, J.L. Persistent ross river virus infection of murine macrophages: An in vitro model for the study of viral relapse and immune modulation during long-term infection. Virology 2002, 301, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, I.; Pan, J.; Takizawa, T.; Nakanishi, Y. Virus clearance through apoptosis-dependent phagocytosis of influenza a virus-infected cells by macrophages. J. Virol. 2000, 74, 3399–3403. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, E.; van Woensel, J.B.; Bem, R.A. Apoptosis in pneumovirus infection. Viruses 2013, 5, 406–422. [Google Scholar] [CrossRef] [PubMed]
- Deszcz, L.; Cencic, R.; Sousa, C.; Kuechler, E.; Skern, T. An antiviral peptide inhibitor that is active against picornavirus 2A proteinases but not cellular caspases. J. Virol. 2006, 80, 9619–9627. [Google Scholar] [CrossRef] [PubMed]
- Deszcz, L.; Seipelt, J.; Vassilieva, E.; Roetzer, A.; Kuechler, E. Antiviral activity of caspase inhibitors: Effect on picornaviral 2A proteinase. FEBS Lett. 2004, 560, 51–55. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, J.A.; Kim, K.H. Effects of a broad-spectrum caspase inhibitor, z-vad(ome)-fmk, on viral hemorrhagic septicemia virus (VHSV) infection-mediated apoptosis and viral replication. Fish Shellfish Immunol. 2016, 51, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Martin, U.; Jarasch, N.; Nestler, M.; Rassmann, A.; Munder, T.; Seitz, S.; Zell, R.; Wutzler, P.; Henke, A. Antiviral effects of pan-caspase inhibitors on the replication of coxsackievirus B3. Apoptosi 2007, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.A. Viruses and the TNF-related cytokines, an evolving battle. Cytokine Growth Factor Rev. 2003, 14, 349–357. [Google Scholar] [CrossRef]
- Mogensen, T.H.; Paludan, S.R. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 2001, 65, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cytokines acting on or secreted by macrophages during intracellular infection (IL-10, IL-12, IFN-gamma). Curr. Opin. Immunol. 1997, 9, 17–23. [Google Scholar] [CrossRef]
- Redpath, S.; Ghazal, P.; Gascoigne, N.R. Hijacking and exploitation of IL-10 by intracellular pathogens. Trends Microbiol. 2001, 9, 86–92. [Google Scholar] [CrossRef]
- Pratheek, B.M.; Suryawanshi, A.R.; Chattopadhyay, S.; Chattopadhyay, S. In silico analysis of MHC-I restricted epitopes of Chikungunya virus proteins: Implication in understanding anti-CHIKV CD8+ T cell response and advancement of epitope based immunotherapy for CHIKV infection. Infect. Genet. Evol. 2015, 31, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, G.V.; Li, Y.; Rassidakis, G.Z.; Medeiros, L.J.; Younes, A. The Hsp90 inhibitor 17-AAG synergizes with doxorubicin and U0126 in anaplastic large cell lymphoma irrespective of Alk expression. Exp. Hematol. 2006, 34, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Nimmanapalli, R.; O’Bryan, E.; Kuhn, D.; Yamaguchi, H.; Wang, H.G.; Bhalla, K.N. Regulation of 17-AAG-induced apoptosis: Role of Bcl-2, Bcl-XL, and bax downstream of 17-AAG-mediated down-regulation of Akt, Raf-1, and Src kinases. Blood 2003, 102, 269–275. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayak, T.K.; Mamidi, P.; Kumar, A.; Singh, L.P.K.; Sahoo, S.S.; Chattopadhyay, S.; Chattopadhyay, S. Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages. Viruses 2017, 9, 3. https://doi.org/10.3390/v9010003
Nayak TK, Mamidi P, Kumar A, Singh LPK, Sahoo SS, Chattopadhyay S, Chattopadhyay S. Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages. Viruses. 2017; 9(1):3. https://doi.org/10.3390/v9010003
Chicago/Turabian StyleNayak, Tapas K., Prabhudutta Mamidi, Abhishek Kumar, Laishram Pradeep K. Singh, Subhransu S. Sahoo, Soma Chattopadhyay, and Subhasis Chattopadhyay. 2017. "Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages" Viruses 9, no. 1: 3. https://doi.org/10.3390/v9010003
APA StyleNayak, T. K., Mamidi, P., Kumar, A., Singh, L. P. K., Sahoo, S. S., Chattopadhyay, S., & Chattopadhyay, S. (2017). Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages. Viruses, 9(1), 3. https://doi.org/10.3390/v9010003